Use the best FRET method for your application.
Dr. Chris Baumann, MAG Biosystems
Förster resonance energy transfer (FRET) has become a common technique in fluorescence microscopy. For FRET to occur, two fluorochromes must be within a defined distance of one another (typically <10 nm) and have their dipoles correctly oriented. Under these conditions, when one fluorochrome (donor) is excited, its excited-state energy can be transferred to the second fluorochrome (acceptor) via a nonradiative transfer. Under ideal conditions — which rarely, if ever, exist — the excitation will selectively excite the donor, and the emission profiles of the donor and acceptor will not overlap.
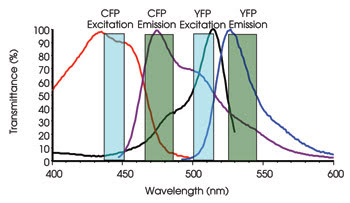
Excitation and emission profiles of CFP and YFP, a popular FRET pair, are shown. Notice that there is significant overlap of the CFP emission into the YFP emission channel (donor bleedthrough).
In reality, the broad spectral properties of the fluorescent probes that are commonly used for FRET are far from ideal (see Figure). An additional complication is that the stoichiometry between the donor and acceptor probes is not always 1:1. As a result, several methods have been developed to separate the actual FRET signal from the noise. These include direct emission ratios, sensitized emission, acceptor photobleaching and polarized FRET experiments.
Intramolecular FRET
FRET experiments can be divided broadly into two classes. The first type, called intramolecular FRET, occurs when the donor and acceptor are linked to a single moiety, and changes within this moiety result in a change in FRET. These types of constructs have been used as small ion sensors for Ca2+ and cAMP monitors of caspase activity, among others.
To monitor intramolecular FRET, you simply excite the donor and calculate the intensity ratio of the donor and acceptor emissions. Because the stoichiometry of the two probes is always 1:1, the amount of direct excitation of the acceptor and donor emission bleedthrough is constant. Therefore, any change in the resulting emission ratio will reflect changes in the level of FRET. Often, these sorts of analyses are performed using a filter wheel to select the emission wavelengths sequentially. Because the ratio typically is determined on a pixel-by-pixel basis, any intracellular movement that occurs between the two sequential images could introduce an error into the analysis. This inaccuracy can be eliminated by using a device such as the MAG Biosystems DV2 and DC2, which allow the two emission channels to be acquired simultaneously.
Intermolecular FRET
The other class of FRET, intermolecular, occurs when the donor and acceptor probes are attached to independent molecules and are brought together via intermolecular interactions. In this case, the stoichiometry of the two probes can vary at each point in the cell. Even without FRET occurring, significant changes in the donor-to-acceptor ratio across the cell are possible, precluding a simple emissions ratio analysis. An attempt to overcome these complications, often called sensitized emission, involves acquiring a series of correction images both with individually labeled and dual-labeled cells. Although this approach can be effective, it is hampered by the additional noise imparted into the analysis as a result of the level of mathematical manipulations required.
Another method for monitoring FRET is acceptor photobleaching, also known as donor dequenching. During FRET, the excitation energy of the donor is transferred to the acceptor, preventing donor emission in a process termed “quenching.” In acceptor photobleaching, an intense light source such as a laser is used to selectively photobleach the acceptor molecule while monitoring donor fluorescence. If FRET is occurring, the intensity of donor emission increases as a result of the loss of its FRET acceptor. The downside of this approach is that it cannot be used dynamically because the acceptor photobleaching process is irreversible. However, this method is well suited as a secondary confirmation FRET in conjunction with another FRET protocol. Therefore, having the ability to add a directed laser source to an existing widefield imaging system, such as the MAG Biosystems FRAP-3D, can be useful for this type of technique.
The final FRET method to be discussed is anisotropy FRET. When a fluorescence probe is excited with plane-polarized light, only the molecules whose fluorescence dipoles are in the correct orientation will be excited. When using a fluorescent protein, the rate of rotational diffusion is slow compared with its fluorescence lifetime, resulting in most of the emission being nearly parallel to the plane of excitation. This is referred to as high anisotropy. However, when FRET occurs, some rotation usually exists between the donor and acceptor dipoles, resulting in a loss in anisotropy. This change in anisotropy can be monitored by exciting the donor with plane-polarized light and by monitoring the acceptor emission intensity parallel and perpendicular to the plane of excitation. Anisotropy is calculated by the following equation:
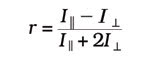
where I|| is the emission intensity parallel to, and I^ is the emission intensity perpendicular to, the excitation polarization. Because fluorescence anisotropy typically is calculated pixel by pixel, intracellular movement occurring between the two sequential images could introduce an error in the analysis. Again, this can be eliminated by using the MAG Biosystems DV2 and DC2.
Meet the author
Chris Baumann is a product manager at MAG Biosystems in Pleasanton, Calif.; e-mail: [email protected].