Veronika Mueller and Christian Eggeling, Max Planck Institute; Håkan Karlsson, Cobolt AB; and Dag Von Gegerfelt, Von Gegerfelt Photonics
Until now, stimulated emission depletion (STED) microscopy typically had been realized with the use of rather large and complex laser systems. But a setup for STED microscopy using a compact and low-noise 0.5-W, 660-nm, single-frequency continuous-wave diode-pumped solid-state (DPSS) laser significantly reduces size, complexity and costs while still providing the same accuracy as a standard STED microscope.
The diffraction of light has put an upper limit on the resolvability of alike objects in far-field fluorescence microscopy, as described by Abbe’s law. In a standard confocal microscope, for example, the diffraction restricts the minimal extent of the excitation laser beam at the sample to about 200 nm in diameter for visible light. Scanning of this laser beam over the sample produces fluorescence images in which details smaller than this minimum spot size appear blurred (Figure 1a).
For decades, this physical limitation has prevented a closer look at cellular structures and therefore concealed many important details of cellular functions. The idea of selectively switching fluorescence on and off has paved the way beyond this limit1 and has brought about a multitude of fluorescence microscopes with nano-meter-scale spatial resolution.2 The very first of these nanoscopy technologies was based on the reversible switching of fluorescence by stimulated emission.1
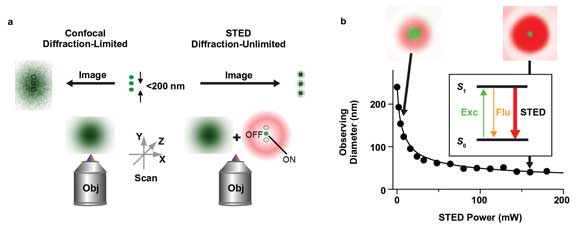
Figure 1. Principle of far-field scanning STED nanoscopy. (a) While the diffraction-limited resolution (>200 nm) of a conventional confocal microscope cannot discern objects closer together than 200 nm (middle), resulting in a blurred image (left), the STED nanoscope features an in principle unlimited spatial resolution by inhibiting fluorescence (OFF) everywhere but at one (or several) spots (ON), revealing nanoscopic details (right). Obj = Objective lens. (b) The resolution of a STED nanoscope increases with the intensity of the STED laser beam: By overlaying the diffraction-limited spot of the excitation laser with the STED laser beam, which features a local intensity zero (red), the diameter of the area in which fluorescence is still allowed (observing diameter, green) shrinks with the STED laser power. Inset: Fluorescence (Flu, orange) is inhibited by de-exciting the fluorescent label’s excited state S1 to the ground state S0 via stimulated emission depletion (STED). Exc = Excitation. All images courtesy of Max Planck Institute for Biophysical Chemistry, Department of Nanobiophotonics (Stefan W. Hell, Göttingen, Germany).
In this stimulated emission depletion (STED) microscopy, the excitation laser spot is overlaid with a redshifted laser beam, which is characterized by one or more zero-intensity points. When the intensity of the latter STED laser light is increased above a certain threshold, fluorescence emission is spatially selectively inhibited, and the area in which fluorescence is still allowed is reduced to a diameter that is much smaller than the diffraction-limited 200 nm (Figure 1). The reduction of the observation volume depends on the intensity of the STED beam and is in principle unlimited. Scanning of such a laser beam configuration can thus produce fluorescence images with an in principle unlimited spatial resolution.2 The technique has proved capable of imaging the molecular distributions inside cells with 20-nm precision and at high speed, and this contributes significantly to the advance of molecular biology research.3 For instance, it allows revealing new insights into the workings of membrane heterogeneity4,5 or activity-dependent organization of neuronal circuits in tissue6 or in vivo.7
A drawback to initial STED microscopy systems was the relatively high level of complexity and the need for large and costly high-power pulsed laser sources, such as a Ti:sapphire pumped ultrafast optical parametric oscillator. But the practical applicability of STED microscopy, especially with regard to imaging inside living cells, has improved substantially over the past years, thanks to the development of new fluorescence labels and new laser technology. For instance, with the right choice of wavelength for the laser beam, STED nanoscopy can be performed on living cells tagged with green fluorescent protein.8
Live-cell STED steps up
Moreover, live-cell STED imaging took a big step forward when scan rates were brought up to several kilohertz, which avoided the buildup of a population of the fluorescent label in the triplet or other (“photo unstable”) dark states.9 This development, combined with the implementation of continuous-wave STED lasers and gated detection schemes, strongly helped to reduce the laser power at the samples and thereby minimize photodamage.10 Furthermore, recent developments in the fluorescence tagging of proteins, for instance the SNAP-tag, have allowed the use of photostable and bright organic dyes inside the living cell (Figure 2a).11
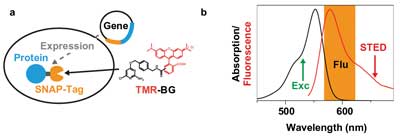
Figure 2. Tetramethylrhodamine (TMR) SNAP-tag. (a) Specific protein labeling inside living cells with TMR is realized by expressing a fusion protein of the protein of interest (blue) and the SNAP-tag (orange) in the cell. On incubation, the functionalized dye TMR-BG (BG = benzylguanine) penetrates inside the cell and binds covalently to the SNAP-tag. (b) Absorption (black) and fluorescence (red) spectra of TMR in water with the excitation wavelength (532 nm, green), the fluorescence detection window (orange) and the optimized STED wavelength at 660 nm.
Now, for the first time, a compact, low-noise CW diode-pumped solid-state (DPSS) red laser has been used for STED microscopy. The use of such lasers substantially reduces the complexity, size and cost of the nanoscope setup. The dye used in the experiments was the membrane-permeable rhodamine dye TMR (tetramethylrhodamine). This dye, which has proved to be useful with the SNAP-tag technology on a STED nanoscope,12 requires a STED laser in the 650- to 670-nm-wavelength range (Figure 2b).
The laser used was a 660-nm DPSS model from Cobolt AB of Sweden; it is based on a miniaturized ring-cavity design and built into a thermocontrolled and hermetically sealed package. The stable output power and low-intensity noise levels enable nanoscopy of TMR-labeled cellular samples; these qualities also permit STED to be combined with single-molecule-based spectroscopic tools such as fluorescence correlation spectroscopy (FCS) for the investigation of nanoscale membrane diffusion of lipids and proteins.
Using a CW laser on a STED microscope, in combination with pulsed excitation and gated detection,13,14 not only eliminates the blurring and the decreased contrast inherent with the CW-STED modality, but also allows live-cell images to be recorded with CW-STED laser powers of around 100 to 150 mW at the sample.14
Recording STED images of TMR-SNAP tagged samples with the gated detection places rather specific demands on the performance of the CW-STED laser. Besides the 650- to 670-nm wavelength already mentioned, a CW-STED light of around 100 to 150 mW is required at the sample, which, because of losses at the different optical parts of the nanoscope, translates into a need for a red laser with at least 400 mW of power.
Another important prerequisite is the use of low-noise lasers, because any fluctuations in the power level compromise the contrast of the final image.14 Figure 3 shows STED images produced using gated detection and a low-noise DPSS laser for STED: the Cobolt Flamenco, which delivers up to 500 mW at 660 nm with an rms noise of less than 0.1 percent.
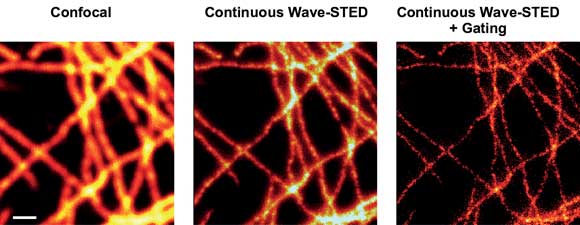
Figure 3. STED imaging of TMR-labeled cells with a 660-nm diode-pumped solid-state laser. Scanning confocal (left) and STED (with [right] and without [middle] gated detection) images of labeled microtubule in mammalian cells. Scale bar: 500 nm.
Excellent resolution
STED images recorded with power of 130 mW at the sample clearly show superior resolution over the diffraction-limited confocal recordings, as exemplified for microtubule of mammalian cells labeled with TMR via the SNAP-tag technology.12 Excitation was performed by a 532-nm pulsed laser system from Picoquant GmbH. Consequently, using a small CW DPSS laser significantly reduces the cost and complexity of a STED nanoscope setup without compromising its performance.
A prominent topic in cellular signaling is the role of heterogeneous organization and the dynamics of molecules in the plasma membrane. It is well known that membrane heterogeneity imposed by lipid-lipid and lipid-protein interactions is involved in membrane-associated processes15 (Figure 4a). These interactions often occur on very small spatial scales of less than 200 nm. Thus, their direct and noninvasive observation in the living cell is fundamentally impeded by the resolution limit of a conventional far-field microscope.
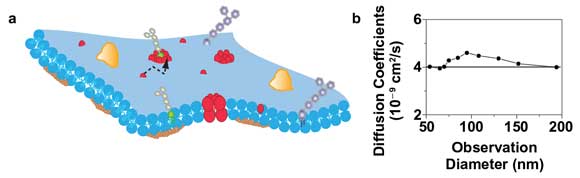
Figure 4. STED-FCS of membrane dynamics with a 660-nm diode-pumped solid-state laser. (a) Scheme of the cellular plasma membrane, where lipids may diffuse (dotted line) heterogeneously due to interactions with other molecules. (b) Diffusion coefficient of a TMR-labeled phosphoethanolamine lipid in the plasma membrane of live mammalian cells determined from FCS data recorded for different sizes of the observation area. The constant values indicate free Brownian diffusion on all spatial scales with a diffusion coefficient of about 0.4 µm2/s (solid line).
Combining STED and FCS
With the superior spatial resolution of STED far-field microscopy, it is possible to directly resolve such membrane heterogeneities. Besides delivering the required spatial resolution to image protein nano-clusters,4 for example, STED nanoscopy can be combined with FCS to disclose complex dynamical processes at the nanoscale.16 In FCS, fluctuations in fluorescence intensity over time are analyzed as labeled molecules move in and out of the measurement volume. STED-FCS enables observation of these molecules’ interactions on nanoscale levels, because slight hindrances in diffusion – e.g., due to heterogeneity and molecular interactions – result in a detectable variation of the transit times through the tiny observation areas. For example, previous STED-FCS experiments showed the potential of observing differences between free and hindered motion of fluorescently labeled lipid molecules. While certain sphingolipid analogs were transiently (~10 ms) trapped on the nanoscale level in cholesterol-mediated molecular complexes, others, such as phosphoglycero-lipid analogs, diffused freely.5
The use of gated detection and CW-STED lasers has proved advantageous for STED-FCS as well as imaging – and for similar reasons.14 STED-FCS in particular requires the use of very low noise lasers because it measures fluctuations in the fluorescence signal; these fluctuations become less detectable for increasing levels of noise. Therefore, we again implemented the 660-nm, low-noise DPSS laser to highlight STED-FCS measurements of a TMR-labeled phospholipid in the plasma membrane of live mammalian cells.
Figure 4b shows results of a STED-FCS study of this diffusion for observation areas of different sizes (created by both increasing the STED power and adjusting the timing of the gated detection14). An accurate observation of the lipid diffusion was possible in this case for observation areas down to 50 nm in diameter. From the transit times and the size of the respective observation areas, we can calculate the apparent diffusion coefficients of the lipids for the different recordings. The independence on the area’s size indicates free Brownian diffusion as expected for these kinds of phospholipids.5,17 The combination of 532-nm excitation and 660-nm STED would also allow the use of the TMR-SNAP-tag for STED-FCS. This is of special interest for the study of membrane heterogeneity because the SNAP-tag would allow labeling of (trans)membrane proteins from both its extracellular and cytosolic parts, which might minimize an alteration of their functionality from the tagging.
Meet the authors
Veronika Mueller is a PhD at Max Planck Institute for Biophysical Chemistry, Department of Nanobiophotonics, Göttingen, Germany; email: [email protected]. Christian Eggeling is a scientist in the same department; email: [email protected]. Håkan Karlsson is CEO of Cobolt AB in Stockholm; email: [email protected]. Dag von Gegerfelt is president of von Gegerfelt Photonics in Bensheim, Germany; email: [email protected].
References
1. S.W. Hell and J. Wichmann (1994). Breaking the diffraction resolution limit by stimulated-emission - stimulated-emission-depletion fluorescence microscopy. Opt Lett, Vol. 19, pp. 780-782.
2. S.W. Hell (2009). Microscopy and its focal switch. Nat Methods, Vol. 6, pp. 24-32.
3. G. Donnert et al (2006). Macromolecular-scale resolution in biological fluorescence microscopy. PNAS, Vol. 103, pp. 11440-11445.
4. J.J. Sieber et al (2007). Anatomy and dynamics of a supramolecular membrane protein cluster. Science, Vol. 317, pp. 1072-1076.
5. C. Eggeling et al (2009). Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature, Vol. 457, pp. 1159-U1121.
6. U.V. Nagerl et al (2008). Live-cell imaging of dendritic spines by STED microscopy. PNAS, Vol. 105, pp. 18982-18987.
7. S. Berning et al (2012). Nanoscopy in a living mouse brain. Science, Vol. 335, p. 551.
8. B.R. Rankin et al (2011). Nanoscopy in a living multicellular organism expressing GFP. Biophys J, Vol. 100, pp. L63–L65.
9. G. Donnert et al (2007). Major signal increase in fluorescence microscopy through dark-state relaxation. Nat Methods, Vol. 4, pp. 81-86.
10. G. Moneron et al (2010). Fast STED microscopy with continuous wave fiber lasers. Opt Express, Vol. 18, pp. 1302-1309.
11. A. Keppler et al (2003). A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol, Vol. 21, pp. 86-89.
12. B. Hein et al (2010). Stimulated emission depletion nanoscopy of living cells using SNAP-tag fusion proteins. Biophys J, Vol. 98, pp. 158-163.
13. J.R. Moffitt et al (2011). Time-gating improves the spatial resolution of STED microscopy. Opt Express, Vol. 19, pp. 4242-4254.
14. G. Vicidomini et al (2011). Sharper low-power STED nanoscopy by time gating. Nat Methods, Vol. 8, pp. 571-573.
15. D. Lingwood and K. Simons (2010). Lipid rafts as a membrane-organizing principle. Science, Vol. 327, pp. 46-50.
16. C. Ringemann et al (2009). Exploring single-molecule dynamics with fluorescence nanoscopy. New J Phys, Vol. 11, p. 103054.
17. V. Mueller et al (2011). STED nanoscopy reveals molecular details of cholesterol- and cytoskeleton-modulated lipid interactions in living cells. Biophys J, Vol. 101, pp. 1651-1660.