Researchers are working to improve FRET, TIRF, FLIM, FRAP and other microscopy methods to get the most accurate data possible.
This may be an obvious statement, but live-cell microscopy is an important, even integral, tool in biomedical research. In studies of cell biology, neurobiology, pharmacology and more, it has enabled insights that would not have been possible by imaging fixed cells. It has allowed investigators to observe changes in cell structures over time as well as the ways in which they interact with other cells, proteins and so on – and thus helped to address a wide array of questions.
Live-cell microscopy encompasses a range of imaging techniques with well-known acronyms: total internal reflection fluorescence (TIRF) microscopy, fluorescence lifetime imaging microscopy (FLIM), Förster resonance energy transfer (FRET) and fluorescence recovery after photobleaching (FRAP). Each of these offers its own strengths for imaging live cells. And each, of course, comes with its own challenges.
Researchers are working to address these challenges. For instance, considerable attention has been paid in recent years to TIRF microscopy, which takes advantage of the total internal reflection phenomenon that occurs when light passes from a high-refractive medium such as a glass coverslip to a low-refractive one – cells or water, for example – to image fluorophores in the near-membrane region of live or fixed cells.
TIRF offers a relatively straightforward means of measuring the intensity of the fluorophores. The challenge is in interpreting what you have measured, according to Sanford Simon, a researcher at Rockefeller University in New York City.
Here, intensity is a function not only of the number of fluorophores but also of the distance between the fluorophore and the coverslip, and of the fluorophore’s orientation relative to that of the excitatory field. “If the molecule rotates slightly,” he said, “it can produce a significant change in intensity. So interpreting the results is not always as straightforward as we’d like it to be.”
To make things more complex, oftentimes the quality of the excitatory field is not as uniform as researchers would like, or the researchers do not have as much control as they would like over the polarization of the excitatory field.
“We have a real need to try to gain control of these things,” Simon said. If researchers were able to control orientation and polarization, for example, they could begin to ask questions such as, “To what extent are changes in intensity due to changes in orientation, relative to changes in other parameters?”
“It sounds like it’s an arcane issue, but it ends up being very important,” Simon added. “By controlling the orientation, for instance, we can get information about what’s happening to the molecules that we couldn’t get before.”
Thus researchers can move beyond the question of “What am I really measuring?” to questions of what is actually happening in cells. In a series of papers starting with one published in the June 2008 issue of Nature, Simon and colleagues have used TIRF and other microscopy techniques to explore the question of where HIV assembles on the cells. “At the time we started the work, it wasn’t resolved whether it assembles on the cell surface or in the internal organelles,” he said. “The best drugs interfere with assembly of the virus, so it’s important to know where this happens.”
Using TIRF, he said, they were able to determine that the virus does in fact assemble on the cell surface.
Quantifying FRET with FLIM
FRET describes the energy transfer between two chromophores – a donor-acceptor pair, in the parlance of the field – and thus can be used to measure distances between the two. Accordingly, scientists have applied it over the decades to studying protein-protein and protein-DNA interactions as well as protein conformational changes, obtaining a wealth of information about in vivo processes.
In more recent years, researchers have been applying FLIM to quantifying FRET in live cells. When detecting protein-protein interactions in these cells, FLIM-based methods are better suited than the more traditional techniques based on steady-state intensity measurements because they allow researchers to determine independently the FRET efficiency and the fraction of the interacting donor – the latter of which, in particular, lets them track the spatiotemporal evolution of the interaction.

Researchers have developed a prototype system called fastFLIM that offers a rapid, user-friendly means of quantifying FRET in live cells using fluorescence lifetime imaging microscopy. Courtesy of Marc Tramier, the National Center for Scientific Research in France.
The FLIM-based method is not without its challenges, however. Most significantly, it is somewhat less than user-friendly, especially when it comes to analyzing and interpreting the results, said Marc Tramier, a researcher with the National Center for Scientific Research in Rennes, France, and the University of Rennes. Also, for live-cell imaging generally, the acquisition speed with FRET is not always high enough for spatiotemporal regulation of protein-protein interaction or biochemical activities.
To address these challenges, Tramier and colleagues – including Sergi Padilla-Parra and Julien Roul, also at the University of Rennes – have developed a prototype system called fastFLIM, based on a white-light laser, a spinning disk, a fast-gated intensified CCD camera and user-friendly software built on the MetaMorph platform. Here, Tramier said, the excitation is optimized to recover fast lifetime images, while the software is especially user-friendly for biologists, in part because it calculates FLIM online without fitting.
The researchers have validated the approach and are preparing a paper about it. They hope that it will help to make FLIM-based FRET available for the broader community of research biologists.
“We have recently used this prototype for FRET biosensors,” Tramier said, “and have concluded that the FLIM quantification is easier than intensity-based methods, even for stoichiometric probes.”
Metal complexes for FLIM and other methods
Interest in live-cell imaging with luminescent metal complexes is on the rise. Using such complexes is attractive for several reasons, said Julia Weinstein of the University of Sheffield in the UK. She recently published a review on this topic with Elizabeth Baggaley, also of the University of Sheffield, and Gareth Williams of the University of Durham. Perhaps the most significant of these: the longer lifetimes offered by the complexes, relative to those of organic fluorophores.
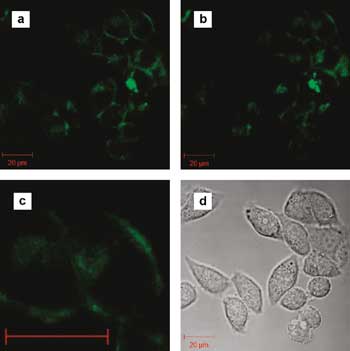
Interest in using luminescent metal complexes for live-cell imaging is growing. Shown here are conventional and two-photon confocal microscopy images of HeLa cells labeled with such complexes (a and b, respectively) as well as the corresponding white-light transmission image (d). Panel c is a magnification of an area in the upper-right corner of panel b showing the membrane. Reprinted with permission from C.-K. Koo, K.-L. Wong, C.W.-Y. Man, H.-L. Tam, S.-W. Tsao, K.-W. Cheah, M.H.-W. Lam, Inorg. Chem. 48 (2009) 7501. ©2009 American Chemical Society.
The lifetime of organic fluorophores is about several nanoseconds, Weinstein said – roughly the same timescale as autofluorescence, making it difficult to separate one from the other. Because the fluorescence in transitional metal complexes emanates from a different state – the triplet state – the lifetimes are considerably longer: hundreds of nanoseconds or even microseconds. “Thus you can gate out the autofluorescence,” she said.
Also, the much longer emission lifetime of transitional metal complexes means that their emission lifetime is considerably more sensitive to the microenvironment – to small molecules such as oxygen or various ions that can be present, for example, in live cells. “This creates unprecedented opportunities for multiplex sensing purely on the basis of the lifetime,” Weinstein said. “This can allow determination, with high precision, of a local concentration of a particular species in question (e.g., ions that regulate a particular physiological process), which underpins, for instance, disease diagnostics and therapeutics development.”

The long lifetimes of metal complexes enable researchers to completely gate out autofluorescence and produce high-contrast, background-free images. Courtesy of Julia Weinstein, the University of Sheffield and Coordination Chemistry Reviews.
Weinstein, Williams and Baggaley are working to develop a time-resolved emission imaging microscopy technique, in collaboration with John Haycock from the University of Sheffield and Stanley Botchway from the Science and Technology Facilities Council of the UK. The technique takes advantage of the long lifetimes of metal complexes to expand the potential of conventional fluorescence lifetime imaging microscopy.