Dr. Orla Hanrahan, Andor Technology
Light-sheet microscopy is faster and less phototoxic than other fluorescence microscopy techniques, making it ideal for studying living organisms and the biological processes that take place within them.
Fluorescence microscopy techniques have become increasingly powerful in terms of resolution, speed and penetration, but most often they only work properly with thin, transparent samples. Whole embryos are fairly opaque, which makes single-cell resolution challenging, especially considering that they are often a few millimeters in size. In addition, the resolution that can be achieved in live specimens is generally lower than in fixed specimens because of a variety of factors: the size of the sample, the scattering of intact and opaque tissue, pigmentation in untreated animals, the movement of living organs (skeletal muscle, gut, heart, blood, eyes, etc.) and the need to keep the sample under physiologically sustainable conditions. These limitations have forced scientists to fix and section their samples, even though many of the highly dynamic processes that occur during development can only be studied in full detail in the intact living embryo.
In recent years, light-sheet microscopy (LSM) has come to the fore as a faster, less phototoxic method than other fluorescence microscopy techniques, making it ideal for studying living organisms and the biological processes that take place within them. With LSM, the illumination is done perpendicularly to the direction of observation. The expanded beam of a laser is focused in only one direction by a cylindrical lens. This way, a thin sheet of light is created in the focal region that can excite fluorescence only in a thin slice (usually a few microns thin) of the sample. Merging a stack of images produces a 3-D image of the sample. Each image in the stack takes a fraction of a second to produce, fast enough to study the beating heart of a zebra fish embryo or to allow images of whole fruit fly embryos (Figure 1) to be generated in just a few seconds. The technique’s speed allows for extended imaging to be carried out on living organisms without any deleterious effects from the dwell time of the laser beam.
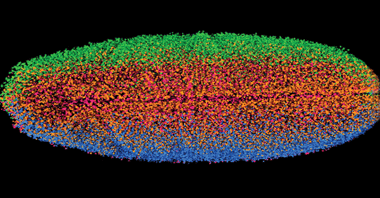
Figure 1. Combining stacks of LSM images into 3-D representations enables researchers to capture images of whole fruit fly embryos in mere seconds. Photo courtesy of Courtesy of Philipp Keller, Janelia Farm Research Campus.
Increasingly advanced versions of LSM have been developed since its introduction over 10 years ago, along with new sample mounting techniques. This is encouraging more and more researchers to use LSM to study a wide range of biological processes.
LSM performs optical sectioning of a specimen with excellent resolution and at high penetration depths, while at the same time being minimally invasive. As the flat, vertical sheet of light slices through the specimen, stimulating fluorescence in a thin, vertical layer, the resultant fluorescence is collected at a horizontal right angle to the direction of the sheet. Unlike conventional fluorescence microscopy, LSM employs two objective lenses: one to focus the light sheet on the specimen and one to collect the fluorescence (Figure 2). This setup has the added benefit that each objective lens can be optimized for a specific role, allowing both an objective lens with a low numerical aperture (NA) to focus the light sheet and a lens with a high NA to collect the fluorescence. The objective lens then collects the fluorescence stimulated in each layer, and a digital camera detects it, allowing a 3-D image of the specimen to be built up layer by layer.
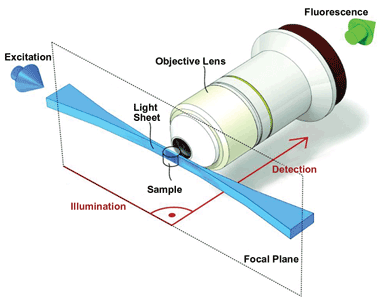
Figure 2. LSM employs two objective lenses: One focuses the light sheet on the specimen, and one collects the fluorescence. Each lens can be optimized for a specific role. Images 2, 3 and 4 courtesy of Development, Vol. 136, pp. 1963-1975 (2009) (doi: 10.1242/dev.022426).
LSM has several important advantages over confocal microscopy (Figure 3), especially for studying living organisms. Most important is that it greatly reduces photobleaching of fluorescent probes and light-induced damage to the specimen. This is because the laser beam is spread out as a sheet rather than being focused onto a small area as a point. Also, no fluorescence is stimulated in regions of the sample not directly exposed to the light sheet. Furthermore, the sheet is applied to each layer of the specimen for a very short period of time, with a single image taking only milliseconds to acquire. Consequently, the specimen isn’t exposed to laser light at a high enough intensity or for a sufficiently long period of time for it to suffer any phototoxic effects or for the fluorescent probes to fade.
Today, single-plane illumination microscopy (SPIM) is one of the most widely used LSM techniques for research. It was first demonstrated by Dr. Ernst Stelzer and colleagues in 2004 to study living fish embryos. SPIM employs all the classic components of a light-sheet microscope and has since formed the blueprint for many subsequent LSM techniques that overcome some of the limitations of the basic version of SPIM. One limitation is the scattering of the light sheet, which gets absorbed and blocked as it passes through the specimen – particularly if it’s a living specimen that cannot be cleared or is not naturally transparent. Furthermore, the stimulated fluorescence is also scattered as it makes its way out of the sample, and the effect worsens deeper inside the sample, producing blurred images. In addition, the presence of opaque compounds within the specimen can completely block portions of the light sheet, creating stripes in the image.
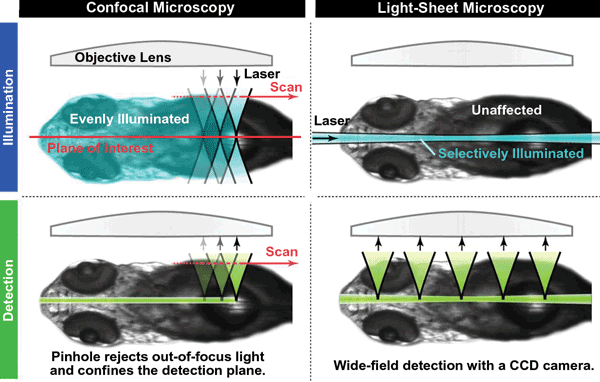
Figure 3. LSM has several advantages over confocal microscopy, especially for studying living organisms.
Using two light sheets to stimulate fluorescence can overcome these scattering problems. In this setup, two lasers are aligned exactly opposite each other on either side of the specimen, ensuring that both sides of the specimen are close to a source of illumination. For the best images, the light sheets are applied successively rather than simultaneously, producing two images that are then merged into a single image. The stripes can be dealt with by applying the light sheet or sheets at various vertical orientations through the same layer of the specimen. As a consequence, the stripes produced by the opaque compounds obscure different parts of the image each time. Merging these different images produces a single image without any stripes. Often, these two techniques are used together, producing a version of SPIM known as multidirectional SPIM (Figure 4).
Another way to deal with the lack of penetration, as well as the scattering of the subsequent fluorescence, is to rotate the sample, taking an image at each rotation, so that the whole specimen is close to the laser and the camera at some point. All the images can then be merged to produce a single, clear image of the specimen. The problem with this approach, known as multiview reconstruction SPIM, is that it takes time to rotate the sample, meaning that it can’t be used to study rapid biological processes; also, it can be difficult to match up the images taken at different positions.

Figure 4. LSM can be combined with other techniques to enable a variety of investigations. OCPI = objective-coupled planar illumination microscopy; mSPIM = multidirectional SPIM; HILO = highly inclined and laminated optical sheet microscopy. obj = objective; s = sample; cyl = cylindrical lens; ch = chamber; det = detection; ill = illumination.
A faster way to do essentially the same thing is to use two lasers and two cameras, with each placed on either side of the specimen in a cross configuration. In this setup, known as multiview SPIM, the two lasers and cameras operate successively, so that one camera collects the fluorescence stimulated first by one light sheet and then the other, and then the other camera does the same. This produces four images that are combined into a single detailed image of a slice through the specimen.
Light sheets produced by a cylindrical lens are associated with certain problems, which researchers are working to fix. The light sheets don’t have a uniform thickness, and they gradually converge to a thinnest point and then widen again. The sample is located at the thinnest point, but if the sample is larger than the extent of the thinnest point, the resolution of the image will be lower at its edges than in the middle.
An alternative method for producing light sheets uses a virtual light sheet. This involves scanning a pencil beam very quickly up and down the sample, building up a light sheet line by line. The exposure time of the camera is matched with the time taken to conduct one scan of the sample, allowing it to collect the same fluorescence as generated by a conventional static sheet and form a single image. This technique produces a thinner sheet with a more uniform thickness. The intensity of the light in the pencil beam needs to be much higher than in a static sheet, around 300 times higher, because each fluorescent probe in the sample is exposed to the beam for only a very short amount of time as it scans down the specimen.
One important advantage of this high intensity is that the beam can be used to conduct two-photon excitation, in which two photons, rather than just one, combine to stimulate fluorescence in a single probe. Two-photon excitation is conducted with beams of infrared light, which is less prone to scattering than visible light and therefore can penetrate farther through tissue. The most advanced version of this technique uses a Bessel beam, which consists of a series of concentric beams that increase in intensity toward the center. Although the entire beam is quite broad, only the central beam, which is both thin (less than 1 μm) and uniform, is sufficiently intense to induce two-photon excitation, producing very high resolution images.
LSM systems not only are much cheaper than other modern microscopy instruments, but they are also much more flexible. Converting a conventional LSM system into a multiview LSM system requires merely purchasing and aligning another laser and digital camera. The system can also be readily updated as existing cameras and lasers can be replaced with newer, more advanced versions. For LSM, the cameras need to be as fast as possible with the ability to record detailed images over a large field of view, because LSM works best using detection lenses with a low magnification and a long focal length.
Manufacturers of scientific cameras are now beginning to take LSM requirements into consideration when developing new models, which will help researchers advance their work and the technology itself.
Meet the author
Dr. Orla Hanrahan is an application specialist in the life sciences at Andor Technology in Belfast; email: [email protected].
DIY LSM
Building your own LSM is easy and cheap: All you need is a laser, a digital camera, the appropriate lenses and a flat surface, in which case a working system can be constructed in just 30 minutes. Indeed, there is a website dedicated to building your own LSM system: OpenSPIM (http://openspim.org). The site provides detailed instructions on constructing an LSM system, as well as open-source software for controlling the system and collecting the images.