Novel probes offer excellent brightness and long life in cellular applications.
Lynn M. Savage, Associate News Editor
Where would modern biomedical research be today without fluorescent
probes? Some would argue that it would be blindly groping down a dark alley, with
no efficient means of discovering the finely honed interplay of proteins and other
substances in the human body, especially at the cellular and molecular levels.
But such probes do exist, whether as dyelike molecules,
artificial metallic dots or a number of other brightly glowing forms. Still, there
is room for improvement among the probes — more brightness and greater endurance,
for example — and there remain biological applications that demand fluorescing
particles that do not yet exist. Investigators across the globe are attempting to
fill these gaps.
Don’t spare the rods
Zinc oxide has several desirable optical properties
— including its high stability in cellular environments — but despite
its advantages, it has received little attention as a potential biosensing material.
However, researchers at Pennsylvania State University in University Park report
in the April 26 ASAP edition of Langmuir that they have constructed substrates
featuring nanoscale ZnO rods that greatly enhance biomolecular fluorescence.
The investigators — Adam Dorfman,
Nitin Kumar and Jong-in Hahm — produced the ZnO nanorods on silicon wafers
by chemical vapor deposition. Using a scanning electron microscope made by FEI Co.
of Hillsboro, Ore., they measured the rods’ average diameter to be 677 ±65
nm. Likewise, they made substrates featuring silicon nanorods with diameters of
547 ±17 nm with which to compare the ZnO rods.
On various substrates, such as glass
and quartz slides, silicon and polymethylmethacrylate (PMMA), the scientists placed
antibovine immunoglobulin conjugated with fluorescein isothiocyanate (FITC-antiIgG).
With a confocal laser scanning microscope from Olympus and a 488-nm argon-ion laser
operating at 40 mW, they confirmed that FITC-antiIgG alone does not fluoresce. Replacing
the substrates with those featuring silicon nanorods provided a negligible amount
of fluorescence, but employing those with ZnO rods resulted in three orders of magnitude
greater fluorescence at the same protein concentration (200 μg/ml) (Figure
1).
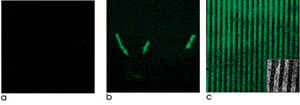
Figure 1. Zinc-oxide nanorods laid out on silicon
scaffolds enhance fluorescence detection. Fluorescein conjugated with an antibody
and placed on silicon wafers or other materials exhibits little fluorescence contrast
(a). However, the same conjugate, when adsorbed onto single ZnO nanorods (b) or
patterned arrays of ZnO nanoplatforms (c), exhibits strong fluorescence. Reprinted
with permission of the American Chemical Society.
They also compared platforms with ZnO
thin films and with ZnO nanorods. They found that, although the film was better
than the other substrate materials, using the rods provided better fluorescence
enhancement.
To evaluate how the ZnO rods might
be used to detect biological agents, the researchers tested them with oligonucleotides
specific to Bacillus anthracis, B. cereus and a modified form of B.
anthracis that carried 6-carboxyfluorescein. No fluorescence was detected from
samples with the B. cereus oligonucleotide, but intense fluorescence was
detected from those with B. anthracis and complementary sequences. They
report that the technique enables the detection of labeled DNA at concentrations
as low as tens of attomoles per liter without amplification.
They note that they are investigating
the exact mechanism behind the enhanced fluorescence.
Reducing the quench
Fluorescent dyes such as Cy5 are handy for cell
and tissue imaging and can act as reliable sensors of protein interactions. However,
you cannot increase the sensitivity of the imaging system or sensor just by adding
an unlimited quantity of a dye onto an antibody or other carrier: Nonfluorescent
dimers form, quenching the fluorescence of the system.
Now, though, investigators at the US
Naval Research Laboratory in Washington, at Scripps Research Institute in La Jolla,
Calif., and at Nova Research Inc. in Alexandria, Va., have devised a more effective
approach for accruing dye particles onto a molecule.
They created a scaffold for Cy5 molecules
from a variant of cowpea mosaic virus. The mutant includes inserted cysteine groups
that enable dyes to readily attach to the virus’s protein shell. The location
of the cysteine ensures that the dye particles are spaced far enough apart (6.5
nm) that nonfluorescent dimers cannot form, thus eliminating quenching.
The researchers report in the April
19 issue of Journal of the American Chemical Society that they fashioned
several versions of the scaffold complexes, composed of different dye-per-virus
ratios up to a maximum of 42 dye molecules per virus. They also added NeutrAvidin,
a binding protein from Amersham Bio?sciences Corp. of Piscataway, N.J., that enables
the complex to act as the recognition element of a sensor.
They used a spectrometer from Varian
Inc. of Palo Alto, Calif., to measure the amount of dye and virus in their samples
and a fluorometer from Horiba Jobin Yvon of Edison, N.J., to measure the fluorescence
intensity of the scaffolded Cy5. They found that the fluorescence signal increased
linearly as the amount of dye on each scaffold rose, indicating minimal quenching
(Figure 2).

Figure 2. Using cowpea mosaic virus as a platform for Cy5 enhances fluorescence because
the fluorophore’s molecules can be closely arranged without overpacking to
the point that self-quenching occurs. In a fluorescence microarray application,
nonscaffolded Cy5 (a, b) does not have the same response as Cy5 on the viral platform
(c). Reprinted with permission of the American Chemical Society.
They also compared the complex with
a mixture of Cy5 and nonmutated virus and with free Cy5. They confirmed that Cy5
attaches to the mutated form of the virus but not to the wild type, and that the
complex provides enhanced fluorescence intensity over the dye alone.
A healthy glow into old age
The investigators noted that energy transfer between
the virus and Cy5 may provide some of the fluorescence enhancement but that it does
not account for all of it. Research is under way to elucidate the full cause.
Many commercial fluorophores are water-soluble,
making them suitable for biological imaging applications. However, they tend not
to be very stable, photobleaching after only a few seconds. A group of researchers
has developed a novel dye based on terrylene diimide that is not only water-soluble,
but also highly fluorescent in various environments for as long as minutes at a
time.
Scientists at Ludwig Maximilians Universität
in Munich and at Max Planck Institüt für Polymerforschung in Mainz, both
in Germany, and at the University of Illinois at Urbana-Champaign synthesized and
characterized WS-TDI, a molecule that has a hydrophobic core but also a number of
hydrophilic sulfonic groups that enable water solubility.
They report in the April 19 issue of
Journal of the American Chemical Society that WS-TDI, although water-soluble,
forms self-quenching aggregates in polar solvents but remains in its monomeric form
in such polymers as PMMA and polyvinyl alcohol. Also, adding a surfactant breaks
up the aggregates into monomers, leading to an increase in fluorescence.
WS-TDI absorbs at ~680 nm and
has a maximum emission of ~720 nm. Using an inverted microscope from Carl
Zeiss of Göttingen, Germany, either a 635-nm laser from PicoQuant GmbH of Berlin
or a 633-nm helium-neon laser and an avalanche photodiode from EG&G Optoelectronics
of Vaudreuil, Quebec, Canada, the researchers investigated the photostability of
WS-TDI, finding that it is more photostable than oxazine-1, sulforhodamine-B and
other commonly used water-soluble dyes.
They also found that WS-TDI is less stable than — and emits about one-third the number of photons of — the water-insoluble form of TDI. They note, however, that the lower stability
and emissions are the price to pay for achieving water solubility.
Comparing dye abilities
The investigators compared the ability of WS-TDI,
Alexa 647 and a styryl dye to image living cells. Where the latter fluorophores
began to photobleach three seconds after irradiation from a 633-nm HeNe or 532-nm
Nd:YAG laser, the cells labeled with WS-TDI remained clearly fluorescent after four
minutes (Figure 3).

Figure 3. Cells loaded with Alexa 647/dextran (a), a styryl dye (b)
or a water-soluble form of terrylene diimide (c) show good fluorescence at the start,
but only the latter maintains its intensity for more than a few seconds. Reprinted
with permission of the American Chemical Society.
Cellular biologists interested in the
kinetics of molecular interactions use dual-color fluorescence cross-correlation
spectroscopy to track the movements of two different fluorophores in the same region.
However, to excite the fluorophores, two lasers, usually of different wavelengths,
must be carefully aligned to the same confocal spot. And, although the technique
can be performed with a single laser, crosstalk and unwanted Förster resonance
energy transfer (FRET) between the fluorescence particles typically occurs.
To improve the technique, researchers
at Riken in Wako and at Hokkaido University in Sapporo, both in Japan, have developed
a fluorescent protein with a Stokes shift of ~180 nm. Because of this large
shift, they dubbed the protein “Keima,” after the knightlike piece used
in the Japanese game shogi. They report in the May issue of Nature Biotechnology
that the variant, which is based on a mutated nonfluorescent protein taken from
the stony coral Montipora spp., exhibits maximum absorption at 440
nm and maximum emission at 620 nm.
They tested the protein in a single-laser
fluorescence cross-correlation spectroscopy setup, pairing it with cyan fluorescent
protein and exciting both fluorophores with a 458-nm argon-ion laser. They found
that the combination enabled simple but efficient use of the technique because Keima
and CFP have nearly the same excitation spectra and easily separable emissions.
Furthermore, no energy transfer occurred between the two proteins.
The scientists then used the technique
to detect the calcium-dependent association between calmodulin and calmodulin-dependent
kinase I (CaMKI). They created various pairings of the new fluorescent protein and
CFP fused with either calmodulin or CaMKI, excited the fluorophores with the 458-nm
laser and viewed the resulting fluorescence using a Zeiss confocal microscope and
40x, 1.2-NA objective. They found that, in general, the amplitude of the cross-correlation
remained low when each sample lacked calcium but increased when calcium was added.
They also tested Keima’s ability
to aid multicolor cell imaging. Using a Hamamatsu Photonics CCD camera and Keima,
CFP and YFP simultaneously in transfected rat cardiac muscle cells, they observed
the concentration of free calcium ions in the cytosol and the mitochondrial
morphology within the highly motile cells. Similarly, they used two variants of
Keima in conjunction with four other fluorophores to visualize the interactions
among the plasma membrane, endoplasmic reticulum, Golgi apparatus, microtubules,
mitochondria and nucleus of living cells (Figure 4).
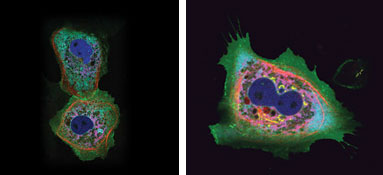
Figure 4. A single 458-nm laser was used to
illuminate cells with up to six different fluorophores, including two variants derived
from the stony coral Montipora spp. Images courtesy of Atsushi Miyawaki of the Riken
Brain Science Institute.
They note that there are chromatic
effects related to the protein’s emissions that must be resolved, but suggest
that the fluorophore and its variants should solve longstanding problems in multicolor
imaging.
When FRET is desired rather than avoided,
enhanced cyan and yellow fluorescent proteins have found widespread use as acceptor-donor
pairs. They also are commonly employed as complementary fluorophores for dual-color
imaging. However, they can be limited by their sensitivity to heat and by their
tendency to aggregate and self-quench.
Enhancing the enhanced
Now researchers at the University of Amsterdam
in the Netherlands have developed optimized variants of enhanced CFP and YFP. These
“superfluorescent proteins” exhibit high brightness compared with their
respective original forms. The investigators created the variants by selective mutation
starting from Venus, another alternate form of YFP that folds more quickly and efficiently
than enhanced YFP, and from Cerulean, a bright variant of enhanced CFP.
In the May 3 ASAP edition of Biochemistry,
the scientists report that they tested the brightness of “super” CFP
and YFP in samples of E. coli. Using a fluorescence microplate reader made
by BioTek Instruments Inc. of Winooski, Vt., they found that E. coli expressing
the variant SCFP3A were nine times brighter than bacteria expressing enhanced CFP
and 1.2 times brighter than those expressing Cerulean. Furthermore, bacteria that
expressed super YFP were 12 times brighter than those that expressed enhanced YFP
and about two times brighter than those that expressed Venus.
To test the capabilities of the new
variants, the investigators transfected cells with either their YFP or CFP variants
and imaged the fluorescence with a CCD camera from Roper Scientific of Tucson, Ariz.,
connected to a Leica stereomicroscope. They noted that SCFP3A, which has a quantum
yield of 0.56, fared less well in live mammalian cells than bacteria but was still
1.5 times brighter than enhanced CFP. Similarly, their YFP variant was 1.5 times
brighter than enhanced YFP in mammalian cells.
Because enhanced CFP and enhanced YFP
frequently are used together as acceptor-donor pairs in FRET, the researchers also
tested the new variants to see whether they would be effective for such use. They
used a Zeiss inverted microscope equipped with a frequency-domain fluorescence lifetime
imaging detector containing a modulated image intensifier from Lambert Instruments
of Leutingewolde, the Netherlands, to determine the fluorescence lifetime of the
particles. They excited the super CFP with a helium-cadmium laser from Melles Griot
of Carlsbad, Calif., and the super YFP with an argon-ion laser, also from Melles
Griot.
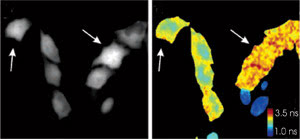
Figure 5. Two variants
of a “super” cyan fluorescent protein cannot be discriminated via their
intensities alone (left, arrows). Because they have different fluorescence lifetimes,
however, phase-lifetime images aid discrimination (right). Reprinted with permission
of the American Chemical Society.
They found that the proteins paired
very well for FRET-based studies because the high quantum yield and extended lifetime
of the cyan variant — combined with the high extinction coefficient of the
yellow variant — result in a marked difference in the CFP fluorescence lifetime
in a FRET and non-FRET situation. They also found that pairing two versions of the
cyan variant — both spectrally the same but one with a much lower lifetime
than the other — permits dual-color imaging while staying in the cyan emission
range (Figure 5).