Neil Anderson, Semrock Inc., and Kengyeh Chu and Jerome Mertz, Boston University
Photobleaching and phototoxicity are unavoidable
in fluorescence microscopy. The phenomenon of photobleaching occurs when, as a result
of photo-induced modifications to the molecular structure, a fluorophore permanently
loses the ability to fluoresce. Phototoxicity arises from photoinduced generation
of singlet oxygen and/or other toxic by-products. Both phenomena are detrimental
to fluorescence microscopy applications, especially in the case of live-cell imaging,
where real-time effects can impair the cell under study. Therefore it is critical
to reduce these consequences to a negligible level. One means of achieving this
reduction without sacrificing image fidelity is to employ a new technique called
active illumination microscopy (AIM). AIM works by maintaining a fixed detection
signal via fast feedback control of the microscope illumination power. Ultimately,
this approach permits fluorescence microscopy with an improved signal-to-noise ratio
(SNR) and little to no image saturation, resulting in clear, crisp high-fidelity
images.
In conventional laser scanning fluorescence microscopy, the laser
source is held at constant power and scanned across the sample, with the emitted
fluorescence signal subsequently measured using a detector, such as a photomultiplier
tube (PMT) or an avalanche photodiode. The signal is assigned a pixel location,
thus allowing one to generate two- and even three-dimensional images with high spatial
resolution. The range of detectable signal levels is defined by the dynamic range.
The dynamic range is most easily understood as being the highest
measurable signal before detector saturation occurs, divided by the smallest detectable
signal. In biological imaging applications, one often encounters problems in acquiring
high-quality fluorescence images on the first attempt without discovering regions
within the field of view that saturate due to higher densities of the target fluorophore.
On the other hand, there may be regions of interest within the same field of view
where low levels of fluorophore exist. The microscopist can deal with the various
levels of fluorescence by acquiring several images with different illumination powers.
Alternatively, this problem can be overcome with AIM. The central
idea behind AIM is to redistribute the dynamic range using real-time feedback to
control the illumination source. By adjusting the output power of a laser on a subpixel
time-scale via feedback control, it is possible to reduce the illumination level
on a real-time basis for bright objects and to increase the illumination level for
faint objects. Because the feedback control responds to the sample conditions, it
eliminates the need for judicious decision making by the microscopist as well as
the need for postprocessing using software. The technique allows high-quality fluorescence
images to be acquired with little to no user intervention.
The AIM approach is shown schematically in Figure 1, where the
dashed-line box encases the feedback control system that is incorporated into a
two-photon microscope platform. The measured fluorescence signal is given by S =
XP2, where X is the measurable parameter of interest and is proportional to the
fluorophore concentration. The feedback loop uses negative feedback to keep the
detected signal at a constant value Sset by modulating the input laser power up
to a maximum power Pmax.
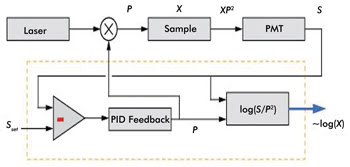
Figure 1. This schematic shows AIM for fluorescence microscopy. The dashed box encases
the integrated AIM feedback control. The fluorescence output XP2 is measured
by a detector, producing an output signal S that is held at a set point Sset
by analog feedback via an electro-optic modulator that controls the illumination
power P. The value X is reconstructed and output on a log scale. PID
= proportional-integral-derivative controller. Adapted from Biomedical Optics
Express by Kengyeh Chu and Jerome Mertz.
When sufficient laser power is available to hold Sset, the system
enters “feedback active” mode. When insufficient laser power is available
to reach Sset, the laser power is automatically set to Pmax, and the system enters
“power limited” mode. In both scenarios, the system evaluates the parameter-of-interest
X and supplies this value as a single output. Although the single-output approach
has benefits from a practical implementation standpoint, it is important to ensure
that the increased dynamic range now obtained is not undermined by the intrinsic
dynamic range of the analog-to-digital conversion. The reconstructed data (i.e.,
X) is thus represented using a logarithmic scale.
This approach may seem foreign to microscopists; however, such
a log scaling approach is ideally suited to AIM, as it captures the characteristics
of good low-signal precision and high overall dynamic range. Whether the data is
represented by a linear or log scale, it can very straightforwardly be converted
a posteriori using appropriate software, if desired.
Fidelity in fluorescence imaging is often corrupted by optical
noise. The benefits of AIM for improving the SNR are shown graphically in Figure
2. Here SNRmin represents a minimum value, due to electrical noise, while SNRmax
represents the maximum value, due to detector saturation. In conventional two-photon
excited fluorescence (TPEF) imaging, the SNR increases proportionately as X1/2.
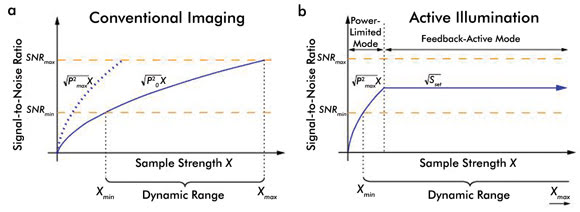
Figure 2. These graphs chart the SNR versus sample strength (X) for (a) conventional
imaging and (b) active illumination microscopy. The SNR follows a standard shot-noise
model in the power-limited regime but is controlled to be at a constant level once
the system reaches the set point Sset in the active-feedback regime.
Adapted from Biomedical Optics Express by Kengyeh Chu and Jerome Mertz.
Increasing the laser power to the maximum possible level permits
lower values of X to be measured. However, increasing the laser power also lowers
the saturation threshold. As a result, the dynamic range Xmax/Xmin remains constant
regardless of the power setting. This limitation is overcome when AIM is implemented.
In AIM, two distinct operation regimes exist. In the feedback-active mode, imaging
becomes saturation-free, and Xmax becomes quite large. In the power-limited mode,
the system operates similarly to a conventional fluorescence microscope, but with
laser power Pmax rather than P0, thereby allowing the user to measure the smallest
possible Xmin.
AIM may seem to be the ultimate cure for the effects of photobleaching
and phototoxicity; however, both still pose a threat, especially if the user does
not fully understand the pros and cons associated with AIM. When increasing Pmax
to allow measurements of low X values, one should recognize that photobleaching
effects can dominate if Pmax is set too high. Similarly, although higher values
of Sset are desirable in that they lead to an increased SNR and improved image fidelity,
they may come at the price of inducing phototoxic effects within the sample. Ultimately
it is left to the discretion of the microscopist to judiciously select values of
Sset and Pmax that best suit a given application.
The main benefits of AIM are demonstrated with an example of TPEF
microscopy of mouse brain hippocampus labeled with GFP. Saturation-free AIM is performed
using more than 200 mW of laser power at 835 nm, but in the standard TPEF experiments,
90 mW or less must be used to avoid detector saturation. The excitation light is
transmitted using a long-pass dichroic having a cutoff at 695 nm and is focused
onto the sample using a high-numerical-aperture microscope objective.
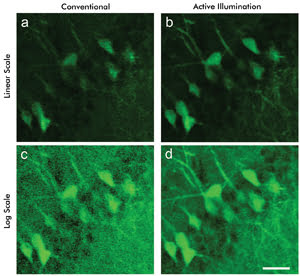
Figure 3. Two-photon emission fluorescence imaging shows a mouse brain stained with GFP.
Conventional TPEF imaging renders the image on a (a) linear and (c) log scale. Imaging
using AIM is (b) linearized in software and (d) acquired on a log scale. Scale bar
20 μm. Courtesy of Kengyeh Chu and Jerome Mertz, Boston University.
The emitted fluorescence, captured by the same microscope objective,
passes through a 60-nm-wide bandpass filter centered at 530 nm before impinging
on the detector, a PMT. Shown in Figures 3a and 3d are two TPEF images acquired
with the microscope operating in standard (linear scale) and AIM (logarithmic scale)
modes. The images are displayed as they were recorded in real time, with no postacquisition
modifications. The application of a simple logarithm (using software) allows one
to convert the linear-scale data shown in Figure 3a to logarithmic-scale data shown
in Figure 3c, or vice versa (Figures 3b and 3d). A direct comparison of the mouse
brain hippocampus images shown in Figures 3a through 3d reveals the main advantage
of AIM: The technique clearly shows weakly fluorescing structures in the sample
that are not visible with conventional TPEF imaging.
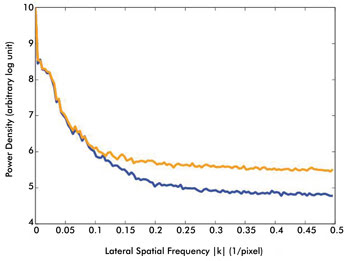
Figure 4. The SNR benefits of AIM are demonstrated in this graph.
Line profiles of the power spectral densities of conventional and AIM TPEF images
are measured from Figure 3a (conventional TPEF imaging, orange trace) and Figure
3b (AIM-TPEF imaging, blue trace). The power density is shown on a log scale, with
the X-axis representing the spatial frequency kx (with ky
set to zero). Operating in AIM mode reduces the noise floor compared to conventional
imaging and directly reflects an improved SNR. Courtesy of Kengyeh Chu and Jerome
Mertz, Boston University.
Improvements in image fidelity (i.e., SNR) afforded by AIM are
demonstrated by plotting the spatial frequency power density of both the conventional
and AIM linear images (Figure 4). From the power spectral density line profiles,
it is clear that the noise floor for the AIM image is reduced compared with that
of the image acquired in conventional TPEF mode.
The benefits derived from applying AIM to low-signal TPEF imaging
are demonstrated in the example above. But AIM also provides benefits for high-signal
imaging, as demonstrated with a sample of fixed mouse brain. When one is operating
in conventional TPEF mode, image saturation is clearly evident in the central region
of the neuron, as indicated by the red arrow in Figure 5a. Switching to AIM avoids
image saturation (See Figure 5b). In addition to saturation-free imaging in regions
where high fluorescence emission is present, AIM also permits imaging of dim features
surrounding the neuron of interest, without sacrificing the SNR.
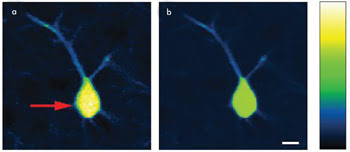
Figure 5. (a) Conventional two-photon excited fluorescence microscopy operating beyond
saturation produced this image of a mouse brain. The red arrow indicates saturation
in the neuron body. (b) In this AIM image of the identical neuron, saturation is
avoided, while the sample strength is measured and displayed. Evident is AIM’s
ability to image dim features without sacrificing the SNR. A nonlinear intensity
scaling is shown on the right to elucidate this effect. Scale bar is 10 μm.
Courtesy of Kengyeh Chu and Jerome Mertz, Boston University.
Conclusion
Active illumination microscopy is a simple and straightforward
means by which to effectively control and manage the effects of photobleaching and
phototoxicity in fluorescence microscopy. Through precise control of the illumination
power, it is possible to eliminate image saturation while enhancing detection sensitivity
at low signal levels. Because AIM requires only simple, straightforward electronics
and only one detection channel, it can easily be employed as a simple “drop
in” addition to any standard one- or two-photon laser scanning fluorescence
microscope platform. Because of this, AIM should provide microscopists with another
powerful method to enhance their imaging tool kit.
Meet the authors
Dr. Neil Anderson is the technology development analyst at Semrock
Inc.; e-mail: [email protected]. Dr. Kengyeh Chu is a postdoctoral researcher;
Chu and professor Jerome Mertz work in the department of biomedical engineering
at Boston University; e-mail: [email protected] and [email protected].
References
1. K.K. Chu et al (October 2007). Enhanced weak-signal sensitivity
in two-photon microscopy by adaptive illumination. Opt Lett, pp. 2846-2848.
2. K.K. Chu et al (August 2010). Practical implementation of log-scale
active illumination microscopy. Biomed Opt Express, pp. 236-245.