Angela Goodacre and Dennis Donley, Olympus America Inc.
Multiphoton microscopy is especially useful when intrinsic fluorescence imaging is combined
with other label-free imaging modalities, such as second-harmonic generation or
coherent anti-Stokes Raman scattering, which may open new windows of opportunity
for research into dynamic cellular events and processes.
The intrinsic fluorescence of cells, their organelles and other biological
elements can be valuable for uncovering new information about tissue function and
cellular processes. In comparison to labeling cells with exogenous dyes that may
alter native physiology or morphology, intrinsic fluorescence, or autofluorescence,
offers unique advantages in studying living cells and tissue. Using intrinsic fluorescence
to image cellular processes in real time allows quantitative evaluation without
perturbing the cell.
The power of imaging intrinsic fluorescence has only recently
begun to be explored as advances in optics and lasers have made multiphoton microscopy
systems more readily available to biological researchers.
Most intrinsically fluorescing constituents absorb light in the
ultraviolet wavelength range and emit in the violet and blue. Because of phototoxicity,
photobleaching and other problems associated with ultraviolet transmission through
optical components, scientists often image intrinsic fluorescence with a multiphoton
excitation system with a pulsed near-infrared laser. In such systems, two or more
long-wavelength photons combine their energies to function as a photon of half the
wavelength. Thus, the fluorescence-excitation effects of two photons of 710 nm
(longer wavelength, which is less damaging to living tissue) are equivalent to a
photon of 355 nm (shorter wavelength, which is usually biologically averse).
In general, the signal from intrinsic fluorescence is much weaker
than from traditional, exogenously induced fluorescence. The two-photon absorption
cross section of nicotinamide adenine dinucleotide (NADH), for instance, is 1 to
0.1 percent the magnitude of conventional fluorophores.1 This means that the multiphoton
laser-scanning microscope in autofluorescence experiments must be optimized for
sensitivity. The system should have transmission characteristics optimized for the
infrared wavelengths of the pulsed laser and dispersion compensation matched to
the exact optical components of the light path.
In highly scattering material such as brain tissue, a tenfold
increase in signal can be attained by maximizing the collection of scattered photons
via external or nondescanned detectors, together with an objective lens that offers
relatively low magnification combined with a high numerical aperture. To ensure
the highest possible excitation efficiency deep within tissue, a correction collar
allows the user to minimize the focal volume.
Two-photon excited intrinsic fluorescence has been reported at
the following wavelengths:
• Reduced forms of nicotinamide adenine dinucleotide including NADH and NADPH, with emission peaks around 450 nm (blue-violet)
• Elastin, retinol and folic acid, with emission peaks around 500 nm (bluish green)
• Riboflavin, with an emission peak at around 540 nm (green)
• Oxidized forms of flavin adenosine dinucleotide, which emit at around 560 to 590 nm (in the yellow range)
• Lignin, a major structural polymer in plant cell walls, which emits a broad spectrum of fluorescence that peaks between 450 and 550 nm.
Multiphoton microscope systems not only facilitate excitation
of ultraviolet-absorbing molecules but also alleviate many of the problems that
arise during excitation and signal collection for autofluorescing specimens because
multiphoton imaging systems, in comparison to their confocal counterparts, rely
on much longer wavelengths of light to induce fluorescence, which are subject to
far less scatter as they pass through tissue.
Multiphoton excitation is limited to a small volume corresponding
to a single point in the image, and photons are delivered in short bursts, or pulses,
lasting for approximately 100 fs. Statistically, the opportunity for two or more
photons to interact with the same fluorophore molecule occurs only at the plane
of focus, where there is a high density of photons, so fluorescence excitation is
effectively limited to the plane of focus, eliminating photobleaching and phototoxicity
in the areas above and below the focal plane.
Redox potential and more
Researchers are using autofluorescence imaging to investigate
oxidative stress, which has been implicated in a wide variety of diseases, including
diabetes, atherosclerosis, neurodegeneration, age-related macular degeneration and
cancer cell pathology. In particular, researchers have found that the reduced forms
of NADH and NADPH exhibit intrinsic blue fluorescence. NADH is a major by-product
of respiration, in contrast to NADPH, which is involved with biosynthesis of lipids
and nucleic acids.
Imaging the fluorescence intensity of NADH and NADPH as a ratio
with the yellow fluorescence of oxidized flavin derivatives can provide a sensitive
means to assess oxidative pathways. The ratio between the two serves as a measure
of the reduction-oxidation capacity of the cell and is known as the cell’s
redox potential, which can affect a range of cellular processes including gene expression,
apoptosis, cancer pathology and enzyme kinetics, allowing the investigation of tissue
alterations due to disease or changes in metabolism.
Dr. Karl Kasischke at the University of Rochester Medical Center
in New York studies the metabolic activity of brain tissue. Most studies of brain
metabolism use cell extracts, so information about the spatial organization and
behavior of different cell types is lost. Kasischke uses the Olympus FV1000MPE multiphoton
microscope system and intrinsic fluorescence to image the pattern of metabolic activity
in relation to cell types in specific regions of the brain and has correlated specific
neural activity with local metabolism. High-resolution microscopy of the mouse cortex
in situ allows his group to map the spatial relationship between blood vessels and
local NADH levels to elucidate mechanisms by which the brain regulates blood flow
to maintain critical oxygen supply.2
Stimulating neural activity causes rapid consumption of glucose,
resulting in increased concentration of tissue NADH. Conversely, the intensity of
NADH fluorescence increases with lower oxygen content in tissue. These findings
suggest NADH imaging as a method to measure the effects of local oxygen tension
during neuronal stimulation and other brain activity. Because the brain contains
86 percent NADH and only 14 percent NADPH, most of the fluorescence measured in
the 420- to 500-nm range is due to NADH. Kasischke’s study reveals defined
regions of low NADH concentration, indicating higher oxygen levels surrounding the
termini of these blood vessels (See figure below). The geometry of these regions suggests
that they represent the limits of oxygen diffusion from blood in arterioles through
tissue to supply neural cells.
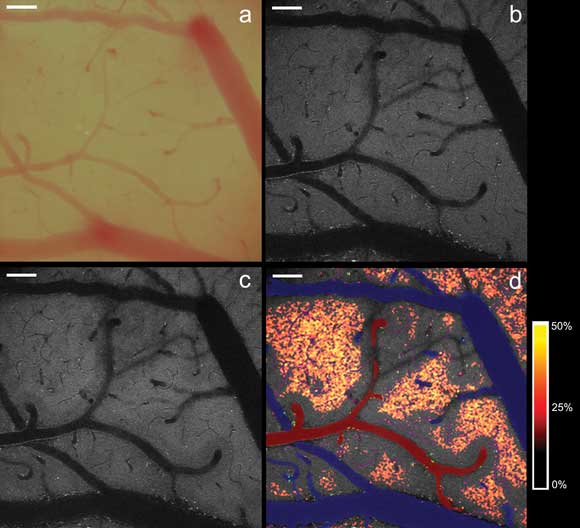
Visualization of microregional hypoxia in the mouse cortex. (a) Exposed mouse cortex as seen under bright-field microscopy. (b) Homogeneous distribution of two-photon excited NADH
fluorescence in the cortex layer under baseline conditions. (c) Heterogeneous distribution
of NADH fluorescence after induction of moderate cortical hypoxia (PaO2 48 mmHg).
(d) Localization of microregional tissue hypoxia. The hypoxia-induced percent increases
in NADH fluorescence (see legend on right) exhibit a distinct geometrical relationship
to the cortical microvasculature (arterioles labeled red, venules labeled blue),
revealing the tissue boundaries of oxygen diffusion from the vasculature. Scale
bars represent 100 μm. (b-d) Images in these panels were captured via intrinsic
fluorescence of the specimen using the Olympus FV1000MPE multiphoton system. Courtesy
of Dr. Karl Kasischke, University of Rochester Medical Center, New York.
Because autofluorescent microscopic imaging of NADH can be used
to localize metabolic activity within subcellular organelles, investigators are
also using it to study mitochondrial anomalies associated with neurodegenerative
diseases, cancer, diabetes and aging – in living cells without destroying
the cells. This is in sharp contrast to traditional investigations of mitochondrial
function, which generally must be performed on extracts and thus lose all information
regarding changes in the number, distribution and morphology of the mitochondria.
Some plant species also exhibit auto-fluorescence, and this characteristic
has helped enable studies in areas such as biofuel production, where the technique
is used to monitor and improve the process of extracting lignin from plant material.
Lignin, a key component of plant cell walls, can make it difficult to procure usable
biomass from readily available plants such as grasses because the lignin blocks
the availability of cellulose, the raw material of biofuels.
By extracting lignin, engineers can make more cellulose available
for fermentation into the sugars that provide energy, but such extraction is notoriously
difficult to achieve. Researchers are now using lignin’s natural autofluorescence
at approximately 530 nm to help measure the lignin content of various plant sources
to help evaluate their potential suitability as biomass for biofuels.
In vivo imaging, as with all imaging of living tissue, presents
enormous challenges. Label-free methodologies are particularly attractive for reasons
beyond the desire to circumvent artifacts caused by reagents. Labeling with a fluorophore
can be prohibitively expensive when the dye must be administered throughout an intact
animal; tissue barriers such as the blood-brain barrier can prevent the label from
reaching its target. Further, the “foreign” nature of fluorophores sometimes
results in their being delivered to the liver or kidneys for excretion, further
complicating experiments.
Meet the authors and acknowledgment
Angela Goodacre is field marketing specialist at Olympus America
Inc.; e-mail: [email protected]. Dennis Donley is marketing business manager,
Scientific Equipment Group, Olympus America Inc.; e-mail: dennis. [email protected].
The authors wish to acknowledge Dr. Karl A. Kasischke, assistant
professor of neurology at the University of Rochester Medical Center, for his support
and assistance in the preparation of this article.
References
1. K.A. Kasischke et al (July 2004). Neural activity triggers
neuronal oxidative metabolism followed by astrocytic glycolysis. Science, pp. 99-103.
2. K.A. Kasischke et al (January 2011). Two-photon NADH imaging
exposes boundaries of oxygen diffusion in cortical vascular supply regions. J Cereb
Blood Flow Metab, pp. 68-81.
Further reading
1. D. Li et al (January 2009). Two-photon autofluorescence microscopy
of multicolor excitation. Opt Lett, pp. 202-204.
2. M. Monici (2005). Cell and tissue autofluorescence research
and diagnostic applications. Biotechnol Ann Rev, Vol. 11, pp. 227-256.
3. W.R. Zipfel et al (June 10, 2003). Live tissue intrinsic emission
microscopy using multiphoton-excited native fluorescence and second harmonic generation.
PNAS, pp. 7075-7080.
4. M.J. Levene et al (April 2004). In vivo multiphoton microscopy
of deep brain tissue. J Neurophysiol, pp. 1908-1912.
5. A.C. Kwan et al (March 2009). Optical visualization of Alzheimer’s
pathology via multiphoton-excited intrinsic fluorescence and second harmonic generation.
Opt Express, pp. 3679-3689.
6. S. Singh et al (September 2009). Visualization of biomass solubilization
and cellulose regeneration during ionic liquid pretreatment of switchgrass. Biotechnol
Bioeng, pp. 68-75.