Marek Scholz, Charles University, and Manjul Shah, Princeton Instruments
A new experimental setup allows researchers to monitor a highly reactive species of molecular oxygen that plays an important role
in a wide range of biological processes.
Oxygen is one of the most important molecules in maintaining life, as well as in mechanisms by which life is extinguished and materials destroyed. For several decades, researchers have been intrigued by the physical and chemical properties of molecular oxygen’s lowest excited state, singlet oxygen (1O2). In particular, singlet oxygen has a unique reactivity that can result in polymer degradation or the death of biological cells. Its role as an intermediate in cell death is exploited by cancer treatments based on photodynamic therapy, a technique in which light is utilized as a medical tool.1,2
In photodynamic therapy, a photosensitizer (PS) is incorporated into abnormal tissues and irradiated with visible light so that it transfers energy to ground-state oxygen via the type II photochemical pathway, producing singlet oxygen (which can be directly detected by its weak 1270-nm emission).3 Owing to the special interest in elucidating the biochemical action of singlet oxygen on the subcellular level, several high-spatial-resolution methods have been proposed to detect 1O2 luminescence using either a single photomultiplier tube, a linear InGaAs detector array or a 2-D InGaAs detector.3
An innovative experimental setup developed and used at Charles University in Prague now permits researchers to perform direct, real-time imaging of singlet oxygen while addressing issues of potential spectral overlap with emitted light from the photosensitizing agent. This fast-data-acquisition microspectroscopy setup relies on the 2-D focal plane array and exceptional NIR sensitivity of a novel InGaAs detector.
The setup has allowed members of the Optical Spectroscopy Group headed by professor Jan Hála to address the issue of potential spectral overlap of 1O2 phosphorescence with the NIR-extended luminescence of the PS, for the first time providing an effective means of distinguishing and separating them.4
Experimental setup for microspectroscopy
The microspectroscopy setup at Charles University uses two detection channels (VIS and NIR) to perform real-time imaging of the very weak NIR phosphorescence of singlet oxygen and photosensitizer simultaneously with the visible fluorescence of the photosensitizer. This setup (Figure 1) enables the acquisition of spectral images based on singlet oxygen and photosensitizer luminescence from individual cells. One dimension of the image is spatial, and the other is spectral, covering a range from 500 to 1700 nm.
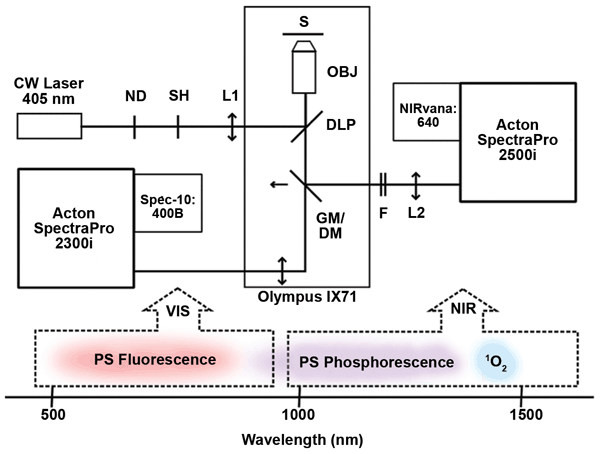
Figure 1. NIR luminescence microspectroscopy setup: Lower portion of diagram depicts spectral regions detected by VIS and NIR paths. Image adapted from Scholz et al (doi: 10.1039/c4pp00121d)4 by permission of the Royal Society of Chemistry (RSC) on behalf of the European Society for Photobiology, the European Photochemistry Association and RSC. ND = neutral density; SH = shutter; S = sample; OBJ = objective; DLP = dichroic longpass mirror; GM = golden mirror; DM = dichroic mirror; F = filter; L1 and L2 = lenses; PS = photosensitizer. Courtesy of Marek Scholz, Charles University.
As illustrated in Figure 1, the real-time imaging setup is built around an inverted fluorescence light microscope (the IX71 from Olympus) and uses a 405-nm CW laser as an excitation source. The laser beam passes through neutral-density filters and is coupled into a NIR-corrected objective by a dichroic longpass mirror. The illuminated spot on the sample (S) is enlarged by inserting a lens (L1) in the excitation path. A golden mirror directs the luminescence emission from the sample, collected by the objective, to a NIR path for detection; removing the golden mirror passes the luminescence emission to a VIS path.
In the NIR path, the signal passes through a combination of NIR longpass and shortpass filters (F) and is focused by an achromatic lens (L2) onto the entrance slit of an imaging spectrograph (an Acton SpectraPro 2500i from Princeton Instruments). The imaging spectrograph is coupled to a NIR-sensitive InGaAs camera (a NIRvana:640 from Princeton Instruments) (Figure 2). The 2-D focal plane array of the InGaAs camera is cooled to −80 °C to reduce dark charge. The spectrograph uses either a grating for spectroscopy or a mirror for imaging. To minimize sample photobleaching, the shutter in the excitation path is controlled by the camera and opened only during exposure times. Alternatively, in the VIS path, a back-illuminated CCD camera (a Spec-10:400B from Princeton Instruments) is coupled to another imaging spectrograph (an Acton SpectraPro 2300i, also from Princeton Instruments).

Figure 2. The NIR path of the experimental setup includes a two-dimensional InGaAs detector coupled to an imaging spectrograph.
The golden mirror in the experimental setup was recently replaced with a shortpass dichroic mirror, so light can now be detected in the VIS and NIR spectral regions simultaneously. The original set of filters has also been modified.4
Results, conclusions
The introduction of the spectral imaging method provides a powerful new tool for distinguishing and separating potential spectral overlap of 1O2 phosphorescence with the NIR-extended luminescence of the photosensitizer. It can be applied to any PS that manifests NIR luminescence. The data presented below illustrates the basic concepts of microscopic spectral imaging; more detail can be found in Scholz et al 2014.4
Figure 3a shows a bright-field image, a VIS fluorescence image and VIS fluorescence spectra of 3T3 mouse fibroblasts that were incubated for 20 hours with 100-μM TMPyP in a D2O-based saline solution. The displayed fluorescence spectra, taken before and after the experiment, reveal a slight change of the spectral shape and fluorescence intensity. The spectra were collected from the region defined by the entrance slit of the imaging spectrograph, represented by the green rectangle in the VIS fluorescence image. The bright-field image demonstrates that the cells are markedly perturbed by a relatively long incubation in D2O. However, this method of incubation leads to increased retention of the PS, which makes it easier to demonstrate several interesting phenomena. For singlet oxygen images from cells using different incubation protocols, refer to Scholz et al 2014.4
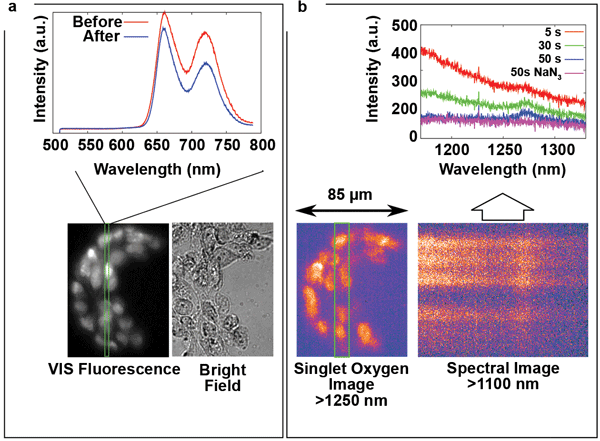
Figure 3. (a) Bright-field image, VIS fluorescence image and VIS fluorescence spectra of 3T3 mouse fibroblasts incubated for 20 h with 100 μM TMPyP in a D2O-based saline solution. The cells are markedly perturbed by a relatively long incubation in D2O. However, this method of incubation leads to increased retention of the PS, which makes it easier to demonstrate several interesting phenomena. For singlet oxygen images from cells using different incubation protocols, please refer to Scholz et al 2014.4 The spectra were collected from the region defined by the entrance slit of the spectrograph (green rectangle). (b) The corresponding singlet-oxygen phosphorescence image and an example of a NIR spectral image, where the vertical dimension is spatial and the horizontal dimension is spectral. The spectral images were collected from the region defined by the entrance slit of the spectrograph (green rectangle). The graphs of the spectra at different time points were obtained by vertical binning of the spectral images. Reproduced from Scholz et al (doi: 10.1039/c4pp00121d)4 by permission of the RSC on behalf of the European Society for Photobiology, the European Photochemistry Association and RSC.

Next, a combination of 850- and 1250-nm longpass filters was used to acquire a 1O2 phosphorescence image with a 10-s exposure. The sample was then left in the dark for 5 minutes, and the liquid surrounding the cells was stirred gently. The broadband background failed to reappear, and there was no change in the sharpness of the NIR or VIS luminescence images (data not shown). The >1250-nm emission intensity even increased slightly, possibly due to recovery of the previously depleted oxygen, indicating both that the signal originated inside the cells and that they did not suffer any substantial PS leakage. Lastly, more evidence that the 1275-nm spectral band can be identified with 1O2 phosphorescence was obtained when this band was quenched by adding 10 mM of NaN3 – a specific 1O2 quencher.
Figure 4 shows that the NIR broadband background luminescence of TMPyP is also evident in bulk solutions of TMPyP in D2O. These solutions (concentration range: 100-500 μM) were placed in a 1-mm-path-length cuvette on the microscope stage for study. Notably, the NIR broadband background was enhanced and the 1O2 phosphorescence was suppressed at strong excitation intensities (9 W/cm2) in a concentrated sample (500 μM of TMPyP), while weaker excitations (2.5 W/cm2) at the same concentration led to a recovery of 1O2 phosphorescence and a decrease in NIR broadband background. The same effect was observed when the concentration was decreased to 100 μM while the excitation intensity was preserved at 9 W/cm2.
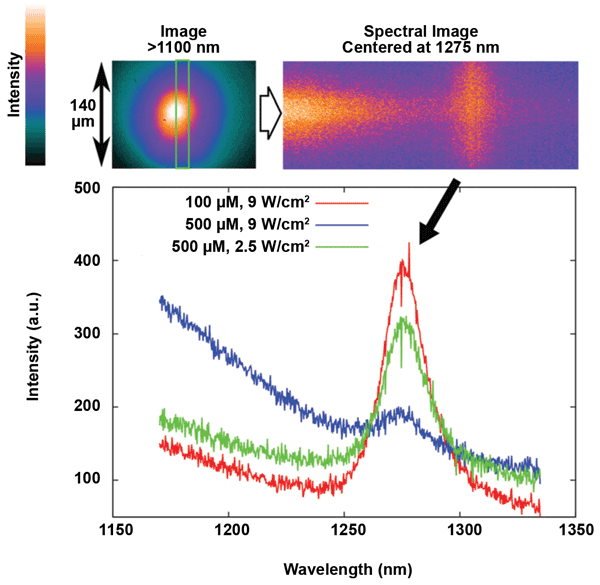
Figure 4. NIR spectra acquired from TMPyP solutions of two different concentrations under diverse excitation powers. A 1-mm cuvette containing the solution was placed on the microscope sample stage. The green rectangle is defined by the entrance slit of the spectrograph and represents the area from which the spectral image was collected. The graph of the spectrum was obtained by vertical binning of the spectral image. Reproduced from Scholz et al (doi: 10.1039/c4pp00121d)4 by permission of the RSC on behalf of the European Society for Photobiology, the European Photochemistry Association and RSC.
The spectral image in Figure 4 (top, right) indicates that the 1170- to 1200-nm emission is limited to the region where excitation is strongest, whereas 1O2 phosphorescence is emitted also from weakly excited regions. It would therefore appear that the NIR broadband background luminescence is enhanced when the concentration of excited states is larger, which in turn implies an excited-state reaction or excited-state complex formation. Each concentration has an optimal excitation intensity that will achieve maximal 1O2-phosphorescence signal and contrast. An excitation power or PS concentration that is too large may actually lead to signal suppression, a cautionary guideline that could also apply to cell samples loaded with TMPyP (in which the local TMPyP concentration can be relatively large). Hence, it is critical to select an optimal excitation intensity for each sample in relation to PS concentration.
The NIR spectral changes observed during cell irradiation in Figure 3b could be partly explained by a drop in the local TMPyP concentration due to photobleaching. However, it is likely that the spectral changes are due to a far more complex interplay involving factors such as formation of TMPyP photoproducts with reduced NIR emission continuing to produce 1O2, or TMPyP relocalization to different cell compartments.
The new microspectroscopy setup, based on a NIR-sensitive 2-D InGaAs focal plane array as a detector, has proved sufficiently sensitive to yield 1O2 images and spectral images of individual D2O-treated fibroblast cells incubated with TMPyP. The setup’s overall efficiency for 1O2 phosphorescence detection was estimated to be 1 to 3 percent. The primary limiting factor was determined to be the numerical aperture of the objective (NA = 0.55).
Meet the authors
Marek Scholz is a Ph.D. student on the faculty of the Department of Chemical Physics and Optics at Charles University in Prague, Czech Republic; email: [email protected]. Manjul Shah is an applications specialist at Princeton Instruments in Trenton, N.J.; email: [email protected].
References
1. C. Schweitzer and R. Schmidt (2003). Physical mechanisms of generation and deactivation of singlet oxygen. Chem Rev, Vol. 103, pp. 1685-1757.
2. E. Skovsen (2004). Progress report: Non-linear two-photon singlet oxygen emission microscopy. Department of Chemistry, University of Aarhus, Denmark.
3. B. Hu et al (2011). NIR area array CCD-based singlet oxygen luminescence imaging for photodynamic therapy. Journal of Physics: Conference Series 277 (doi: 10.1088/1742-6596/277/1/012011).
4. M. Scholz et al (2014). Real-time luminescence microspectroscopy monitoring of singlet oxygen in individual cells. Photochem Photobiol Sci, Vol. 13, pp. 1203-1212 (doi: 10.1039/c4pp00121d).