Dr. Brian Rankin, !%Mobius Photonics%!
Recent advances allow superresolution microscopy to deliver on its promise of using focused light to provide glimpses of the inner workings of living organisms, resolving structures on the scale of tens of nanometers.
The past decade witnessed a renaissance in far-field light microscopy. After initial demonstrations proved that the diffraction-limited resolution barrier could be overcome by manipulating the molecular states of fluorophores used to label the sample, superresolution approaches and methods for their implementation multiplied. The lenses of superresolution microscopes are increasingly directed at the neuron, providing nano-scale images of the delicate machinery of thought itself.
The far-field light microscope is an indispensable observational tool in the sciences. However, although its optical implementation has been continually improved since its invention, the fact remains that the smallest focal spot attainable with visible light is limited by diffraction to ~200 nm. For years, this obscured structures on smaller-distance scales and fueled the development of other types of microscopy – such as the atomic force and electron microscopes – to sidestep the limitation.
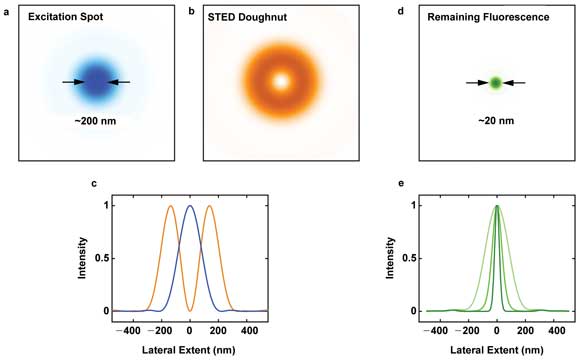
A standard diffraction-limited focal spot (a) is used to excite a fluorophore. The doughnut-shaped STED (stimulated emission depletion) beam (b) with a zero-intensity spot in the center is co-aligned and superimposed on the excitation spot (c) to silence fluorescence at the periphery, resulting in a much smaller area of remaining fluorescence (d). Both beams scan through the sample, point by point, to construct a subdiffraction-resolution image. Increasing the STED intensity increases the fluorescence silencing affected by the STED beam, yielding an ever-smaller spot of remaining fluorescence (e) and higher image resolution. Courtesy of Mobius Photonics.
In the biological sciences, however, using light to scan through the interior of transparent samples (e.g., cells and thin layers of tissue) is hard to beat. Labeling biological structures of interest with fluorophores – using the high specificity of either immunochemistry or genetically encoded fluorescent proteins – enables excellent signal-to-noise ratios when imaging even individual proteins within the highly complex environments of cells or organisms.
Additionally, by using fluorophores to label biological structures of interest using the high specificity of either immunochemistry or genetically encoded fluorescent proteins, excellent signal-to-noise ratios can be obtained when imaging even individual proteins within the highly complex environments of cells or organisms.
Further, by taking into account the photophysics of the fluorophores themselves, a resolution far higher than ~200 nm can be achieved. The physical insight is this: If fluorophores within the same diffraction-limited volume emit their photons at different times, their individual signals can be separated and the locations of closely adjacent fluorophores recorded.1 In other words, by sequentially registering the locations of the fluorophores in time, spectrally identical photons originating from nearby points in the sample can be distinguished from one another.
STED microscopy
Stimulated emission depletion (STED) microscopy achieves time-sequential detection of photons from adjacent subdiffraction-size volumes by using stimulated emission to suppress fluorescence so that the photons from fluorophores closer than ~200 nm can be gathered sequentially.
STED uses two light beams. The first, the excitation beam, is focused to a standard, diffraction-limited spot and excites fluorophores from their ground state into their fluorescent state. The second – the STED beam – is redshifted relative to the excitation beam and has a doughnut-shaped intensity distribution in the focal plane with a spot of zero intensity in the center. Via stimulated emission induced by the photons in the STED beam, fluorescence emission is suppressed everywhere but the center of the doughnut pattern. The image is formed point-by-point by scanning the excitation and STED beams together through the sample. By increasing the intensity of the STED beam, the threshold intensity required to fully suppress fluorescence moves closer to the center; the spot of remaining fluorescence becomes smaller, resulting in a higher resolution for the fluorescence image.
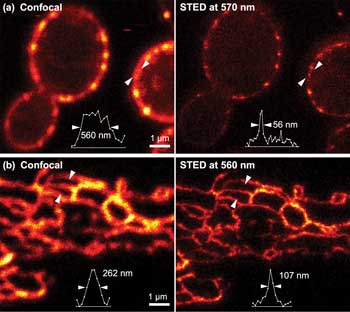
(a) STED image (right) of live yeast cells labeled with GFP, with a standard confocal image (left) for comparison. The pair of arrows in each image indicate the location at which the intensity line profile, shown below in each pane, is taken. The labeled structures are eisosomes, protein assemblies in the cell membrane that define sites for endocytosis. Pulsed 570-nm STED light was used and a resolution higher than 60 nm was obtained. (b) STED image (right) of the endoplasmic reticulum in a live cultured mammalian cell labeled with GFP. Pulsed STED at 560 nm was used. Live cell images courtesy of Stefan Hell, Max Planck Institute for Biophysical Chemistry.
Implementation
Recent advances have dramatically simplified the implementation of STED and extended its utility. Light sources for STED fluorescence suppression must have sufficient intensity for the stimulated emission transition to outcompete fluorescence emission. Also, the light must lie within the spectral range where the cross section for stimulated emission is sufficient for silencing fluorescence emission.
Standard fluorescent proteins including GFP (green), YFP (yellow), RFP (red) and their variants are STED-compatible, allowing researchers to image living samples expressing them with subdiffraction resolution. The STED light required for imaging with such proteins spans the visible spectrum from green to red. Not long ago, the types of suitable lasers available for producing yellow light with sufficient intensity for STED were complicated to operate, requiring STED researchers to also be laser scientists.
For example, the workhorse visible light source for STED has been the Ti:sapphire laser pumping an optical parametric oscillator and requiring pulse stretching from ~200 fs to several hundred picoseconds using dispersive glass rods and optical fiber. New fiber lasers, both pulsed and continuous wave – typically single wavelength but also with tunable or supercontinuum output – have drastically simplified matters. Light suitable for STED microscopy can now be produced with turnkey operation, freeing researchers to devote more time to biological imaging and less to optics. The ideal source produces a broad spectral output with sufficient intensity for STED, giving the user the flexibility to employ a variety of fluorophores – including common fluorescent proteins – with high resolution and high image contrast.
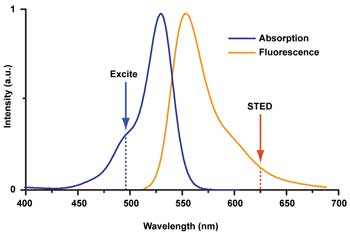
The STED wavelength is tuned to the fluorophore used. As with standard fluorescence microscopy, the fluorophore is excited using a wavelength within the absorption spectrum. The STED wavelength is usually chosen to lie in the tail of the fluorescence emission spectrum, where re-excitation resulting from the STED beam is minimal but the cross section for stimulated emission is sufficient for fluorescence silencing. A tunable STED laser enables different, spectrally distinct fluorophores to be used with the same setup. Courtesy of Mobius Photonics.
Creating doughnut-shaped intensity distributions for the STED beam has been simplified by both commercially available phase masks and birefringent optical elements developed to enable automatically co-aligned excitation and STED beams for easy implementation.2 Beam-scanning STED setups, now commonplace, also have enabled rapid image acquisition of super-resolution movies of moving samples.
Neurobiology at the nanoscale
The area of research where superresolution has perhaps the most to offer is neuroscience. Many interesting neuron features are of dimensions that are at or below the diffraction limit. Aspects of neuron morphology, such as the protrusions from the main cell body, called spines, can be seen only unclearly with conventional light microscopes. The dynamic distribution of synaptic vesicles in the neuron cell body can be obscured because of the close spacing of the vesicles to one another.3
The synaptic cleft – the gap between the pre- and postsynaptic neurons that is flooded with neurotransmitters during cell communication – is a mere few tens of nanometers across. The complex, dynamic spatial distribution of proteins involved in synaptic function on the pre- and postsynaptic sides of the cleft cannot be resolved with standard light microscopes because the spacing between many proteins is also on the subdiffraction distance scale of several tens of nanometers.4
Imaging neurons in living animals
The great advantage of STED microscopy is its ability to create images of these nanoscale neuronal structures within living organisms. Although much can be learned from cultured neurons and brain slices, the way in which a neuron interacts with its neighbors – within the context of the living nervous system – is of great interest. In vivo investigations can provide the most accurate models for nerve cell function, offering the possibility to investigate the nervous system as it processes information and as the organism responds to stimuli.
GFP, YFP and RFP are used ubiquitously as markers in model organisms in the neurosciences to tag structures to visualize their spatial and temporal distribution. STED has matured to a point that it leverages this spectrum of standard endogenous markers and is currently being applied to imaging studies in living organisms that benefit from nanoscale resolution. To date, researchers have used STED to image neurons in two types of live model organisms: the nematode Caenorhabditis elegans and the mouse.
C. elegans
A roundworm ~1 mm in length, C. elegans is a standard model organism for studies in cell development and cell function within the context of a complete organism. Although multicellular, it is composed of ~1000 cells in total, and its nervous system consists of only 302 neurons, a number tractable for complete study. This simplicity allows researchers to study the complete development trajectory of each cell and its function within the organism. C. elegans also is transparent, so every point within the intact worm is accessible with a light microscope.
C. elegans is the first living animal to be imaged with superresolution microscopy.5 Specifically, STED microscopy was used to obtain images of neurons labeled with GFP at subdiffraction resolution, the first demonstration of superresolution imaging in an intact, living, multicellular organism.
In contrast to standard confocal images taken for comparison, STED images revealed a lack of connectivity in branched sections of the neurons. Being able to tell where a nerve cell ends is invaluable in understanding the nuances of its function.
In contrast to the much simpler C. elegans, the mouse possesses a complex neurobiology similar to our own. Using the mouse model to investigate neurobiology comes with additional challenges, however. To state the obvious, mice are large and opaque, necessitating more extensive manipulation than placing them between a slide and cover slip to obtain microscope images of their nervous systems, and movement caused by the heartbeat and respiration can be highly disruptive when imaging at the micron or nanometer scale.
Imaging neurons live
Overcoming these challenges, investigators in Germany recently used STED to obtain images of neurons in the brain of a living mouse.6 Specifically, the study examined the movement of dendritic spines, taking images every 7 to 8 min over a period of half an hour to create a time-lapse record of spine plasticity. These subtle morphological changes in nerve structure are difficult to observe in diffraction-limited images, but were clearly resolved in the STED images.
Dendritic spine plasticity previously had been observed in brain slices of young, developing mice, but not in the brain of mature mice. To enable fluorescence imaging, the mice used were transgenic, expressing EYFP (enhanced yellow fluorescent protein) in the cytoplasm of the neurons of interest. The STED images exhibited resolution better than 70 nm, demonstrating a fourfold improvement in resolution over standard confocal microscope images.
Future of superresolution
For years, superresolution imaging has held promise for leveraging the advantages of focused light to investigate the machinery of life in vivo, in real time, at the nanoscale. This promise is now being fulfilled, and the foray of superresolution microscopy into living organisms has begun. Soon, imaging may be extended to other model systems such as zebra fish and fruit flies.
The field is still developing rapidly, however. There is no doubt that continuing refinements to its technical implementation will further extend its performance and lower the barriers to more widespread implementation.
Meet the author
Dr. Brian Rankin is a scientist at Mobius Photonics Inc. and a graduate of the department of nanobiophotonics at Max Planck Institute for Biophysical Chemistry in Göttingen, Germany; email: [email protected].
References
1. S.W. Hell (2009). Perspective Abstract: Microscopy and its focal switch, Nat Meth, Vol. 6, pp. 24-32.
2. M. Reuss et al (2010). Birefringent device converts a standard scanning microscope into a STED microscope that also maps molecular orientation. Opt Exp, Vol. 18, Issue 2, pp. 1049-1058.
3. V. Westphal et al (2008). Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science, Vol. 320, No. 5873, pp. 246-249.
4. A. Dani et al (2010). Superresolution imaging of chemical synapses in the brain. Neuron, Vol. 68, Issue 5, pp. 843-856.
5. B.R. Rankin et al (2011). Nanoscopy in a living multicellular organism expressing GFP. Biophys J, Vol. 100, Issue 12, pp. L63-L65.
6. S. Berning et al (2012). Nanoscopy in a Living Mouse Brain. Science, Vol. 335, p. 551.