As fluorescence microscopy advances, excitation light source selection becomes more important – and the best choice depends on the application.
To take detailed images of biological structures, fluorescence microscopy relies on intense light. Whether a gas discharge lamp, laser or LED, the light source influences the resolution of the pictures – and any damage that a sample can suffer in the course of imaging.
A strong light source must stimulate fluorescence emission from fluorochromes or fluorophores, which latch onto cells, organelles or even individual molecules, and then respond to a well-defined wavelength of light. When this light hits the fluorochrome, it excites the dye, which releases a similarly specific light signal, albeit one with lower energy than the excitation signal. The emitted light then returns to the detector through a filter, providing a picture of the fluorochrome-marked sample. Various fluorochromes bond to different biological structures and require a different range of wavelengths to stimulate fluorescence.
 To achieve the best possible image, the incoming light must be of a high intensity, to stimulate as much emission as possible. But it must minimize any potential light damage to the sample as well. So, instead of flooding the field with an entire spectrum, the excitation signal should contain only those wavelengths that trigger fluorescence from the fluorochromes in use.
To achieve the best possible image, the incoming light must be of a high intensity, to stimulate as much emission as possible. But it must minimize any potential light damage to the sample as well. So, instead of flooding the field with an entire spectrum, the excitation signal should contain only those wavelengths that trigger fluorescence from the fluorochromes in use.
Lamp illumination
The original fluorescence microscopes relied on gas discharge lamps, also called arc or burner lamps. These light sources provide excellent wide-field illumination and are still in use today. Burner lamps contain gases such as xenon, vaporized mercury or metal halides maintained at a high pressure between two electrodes. When an electrical signal passes between the electrodes, it ionizes the gas in an arc that creates both heat and high-intensity light. Because these lamps produce a wide range of wavelengths from the UV to the IR, an excitation filter reduces the light to a more manageable range.
Of the various gases available, mercury is the most common, followed by xenon. Mercury burners are intense, but the intensity is not consistent across the entire spectrum and peaks at specific wavelengths, particularly in the UV range. Xenon arc lamps have a more consistent intensity across the entire spectrum – although this intensity is higher in the IR range – and thus produce a great deal of heat, which can also harm live samples.
The ideal excitation wavelength will depend on the type of fluorochrome used to stain the sample. An excitation filter can select only the desired wavelengths, but it does not completely suppress all of the peripheral light. For best performance, the light source should have a high intensity in the fluorochrome’s excitation range, which means that its emission spectrum must be taken into account.
The variable intensity of arc lamps deteriorates over time. By the time their bulbs reach the ends of their limited lifetimes – mercury bulbs last about 200 hours and xenon ones hold out up to 1200 hours – their emission spectra will have changed, which may require recalibration. And gas discharge lamps cannot turn instantly on or off to preserve their working lives. Because they require some time to warm up before reaching peak intensity, they are generally turned on for continuous periods of time.
Gas discharge lamps’ inability to switch quickly between off and on makes imaging live samples difficult. When living samples are overexposed to high-intensity light for a long period, phototoxicity and cell death can set in; even fixed samples might suffer from photobleaching. To prevent this, a filter can reduce the intensity of the light, or exposure time can be limited – or an alternative light source might come into play.
“LEDs are becoming more and more common,” said Kristen F. Orlowski, product marketing manager for light microscopy at the US arm of optical systems manufacturer Carl Zeiss Microscopy LLC in Thornwood, N.Y.

Live green brain coral (Goniastrea sp.) are seen underwater: The purple color is the natural fluorescence of the coral, except for near-violet LED illumination that was used to highlight some nearly transparent tissue. This image took fifth prize in the 2011 Olympus BioScapes imaging competition. Photographer: James Nicholson, NOAA/NOS/NCCOS Center for Coastal Environmental Health & Biomolecular Research, Fort Johnson Marine Lab in Charleston, S.C.
With the development of light-emitting diodes came a source of monochromatic light: Each LED produces a limited range of wavelengths. In these semiconductor light sources, an applied current allows electrons and electron holes to recombine, which produces photons. The energy and wavelength of the photons depend on the size of the energy gap in the semiconductor. This trait allows manufacturers to craft LEDs that produce light in a narrow range of wavelengths and, thus, require less filtering, Orlowski said. Not only can LED light sources do away with filters, but LEDs require less power than arc lamps, last longer and can instantly switch on or off, enabling multiple LED modules to stimulate emission from more than one fluorochrome in a single sample and to create time-lapse images. As an LED ages, the illumination intensity does not fade as it does in an older arc lamp.
For each fluorochrome’s desired wavelength, an LED whose light falls into that range can be manufactured.
But this stable, monochromatic illumination source has one big disadvantage in intensity – or lack thereof. When LEDs were first developed, they had extremely low intensities. They have grown steadily brighter, however, and although this trend is predicted to continue, they still pale beside the intensity of arc lamps. The weaker intensity is great for living cells, but it means that the image resolution suffers. LEDs are finding some use in fluorescence microscopy, but they are not yet the most widely used light source.
Both arc lamps and LEDs illuminate the entire sample at once in the low-resolution technique called wide-field microscopy. Of course, “low resolution” is relative: This method can still capture images of individual cells and organs. Although wide-field microscopy cannot capture three dimensions, computer software can easily assemble optical sections into a 3-D reconstruction.
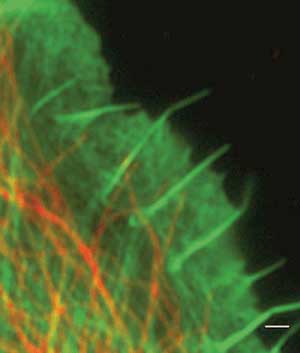
Wide-field microscopy may be relatively low resolution, but it can still image individual cells and organs. Here, a wide-field microscopy image shows actin (green) and tubulin (red) cytoskeleton in a primary chicken fibroblast. Courtesy of Carl Zeiss MicroImaging LLC.
Clearly, wide-field imaging is perfectly sufficient for many researchers’ needs. But to snap higher-resolution images in 3-D, more advanced techniques such as confocal and multiphoton fluorescence microscopy are required. These methods require a more focused light source: lasers.
Laser illumination
Lasers combine some of the best aspects of arc lamps and LEDs. As with lamps, they provide intense light, but because the light is focused on a point rather than over the whole field, the high intensity isn’t as damaging to a living cell. And as with LEDs, lasers are long-lasting and provide light in a specific wavelength range, negating the need for filters.
But while these perks make lasers perhaps the best light source for fluorescence microscopy, they have not quite taken over because of their high price. A fluorescence microscope that uses a laser light source can cost from $20,000 to more than $150,000. In comparison, LEDs range from $10,000 to $20,000, and arc lamps cost a mere $6000 to $8000.
“I would predict that, ultimately, lasers will be used exclusively for all forms of microscopy as soon as their price starts coming down,” said Michael W. Davidson, a scientist at the National High Magnetic Field Laboratory at Florida State University in Tallahassee. “There’s just no reason to use anything else.”
It’s not only their ability to produce intense monochromatic light that makes lasers the best option for fluorescence microscopy. They also are focused, capable of illuminating only a small part of the sample rather than the entire field.
“When you shoot light through the whole sample and only get a little light back, it’s inefficient,” said Scott Olenych, a researcher and product marketing manager at Carl Zeiss MicroImaging LLC, also in Thornwood. It also causes photobleaching. Instead of wasting light on the entire sample, Olenych has worked on a new technique where lasers form a “light sheet,” illuminating the 2-D plane of the sample where the detector is focused. This method is called selective plane illumination.
“When you shoot your light source through your sample, the emitted fluorescence comes from every single focal plane, even the ones not in focus,” Olenych said. “That creates out-of-focus blur. When you have blur, you can’t get a nice sharp view of any of the planes because the light from the other planes obscures the in-focus portion.”
By focusing on a single plane at a time and then reconstructing a 3-D image, the light sheet method avoids photobleaching and is ideal for large, live samples. However, it is not the most common use for laser illumination. More frequently, lasers provide point illumination in confocal, multiphoton and even newer, higher-resolution microscopy techniques.
In wide-field microscopy, the entire sample is illuminated at the same time, so the entire picture hits the detector at once. Selective plane illumination techniques use a laser from the side to form a 2-D plane. And in confocal laser scanning microscopy, a laser illuminates a single point of the sample at a time, and only gradually traces over the entire field. Each point is sent to the detector through a pinhole to eliminate any out-of-focus light, and then reconstructed into a more detailed picture. By illuminating different focal planes of the sample, 3-D images can be constructed as well.
Although the light in confocal microscopy is more focused and thus less harmful to a sample than in a wide-field shot, it can still cause photobleaching and phototoxicity because the light must stay on the sample longer, and it illuminates all planes at once rather than one at a time. Plus, if the material in the sample scatters light easily, the pinhole cannot easily eliminate out-of-focus light.
But in another laser-lit imaging technique, called multiphoton fluorescence microscopy, the light has a longer wavelength and lower energy. To excite each fluorochrome, the fluorescing molecule must be hit by more than one low-energy photon at once. Because the fluorochrome won’t fluoresce without at least a double-photon hit, this technique does not excite fluorescence along the entire path of the laser beam – only at the point where the light is focused. This prevents damage to the sample.
Producing two or more photons at once does require more power from the light source. In this imaging method, laser pulses rather than continuous beams are used, requiring a different type of laser. The lasers adopted for multiphoton microscopy tend to cost more than the instruments used in confocal laser scanning microscopy and to require extra cooling systems to function properly.
As it captures light from deep within a sample while causing minimal harm, multiphoton microscopy is excellent for nabbing pictures of live cells in even a thick multilayer sample. In fact, this type of imaging can be used to map organs in living creatures; e.g., creating pictures of cells in a living brain.
In these high-resolution microscopy methods, as the fluorescing light returns to the detector, it spreads out slightly because of diffraction and limits the possible resolution. This limit used to be considered insurmountable, but in recent years, superresolution microscopy techniques, such as stimulated emission depletion and photoactivated localization, have smashed the diffraction barrier and made extremely detailed images possible.
These pictures have a resolution at the nanoscale and can show organelles and individual molecules in unprecedented detail. In fact, these techniques make it possible to watch molecules moving around a living cell. Superresolution microscopy has taken images of molecules passing across a cell membrane, proteins folding in real time and even HIV particles docking in a cell.
Superresolution images look great, but they aren’t always necessary. Ultimately, the best light source for the job will depend on the needs of the sample – and on the lab’s budget.