Microscopy techniques enable high-speed 3D imaging of biological systems, including cell dynamics and brain network interactions.
Kevin Tsia, University of Hong Kong, Jianglai Wu, University of California, Berkeley, Na Ji, University of California, Berkeley
Ongoing innovations in optical microscopy have fueled life science research, allowing scientists to visualize how complex biological systems work and break down. These developments have required specifications beyond the need for high-resolution views of cells, tissues, organs, and organisms. There is also an ongoing search for new microscopy tools that are fast enough to capture high-speed (submillisecond or shorter) motion pictures of biological dynamics and to have a high enough throughput to interrogate large (millions or beyond) populations of cells for unravelling the intricate cellular heterogeneity. Most importantly, the tools need to be gentle enough to ensure minimal risk of photodamage for monitoring long-term (hours to days) biological processes.
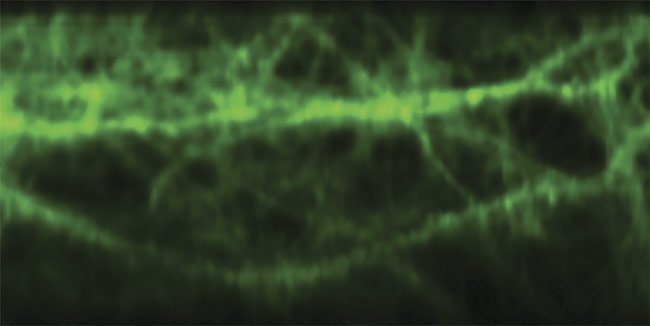
Multiphoton brain imaging, enabled by free-space angular-chirp-enhanced delay (FACED), reveals the signal patterns of glutamate, an excitatory neurotransmitter released by nerve cells in the brain. Courtesy of Kevin Tsia/The University of Hong Kong.
These trends have been the driving force behind the unveiling of many new forms of microscopic imaging and illumination, often enabled by innovative use of components. For example, an emerging approach using the infinity mirror concept has been demonstrated to generate a shower of laser-scanning foci at least 10 to 100× faster than other existing galvanometric (galvo) mirror scanning methods. The technique, called free-space angular-chirp-enhanced delay (FACED)1, essentially resembles the infinity mirror effect, which generates a series of receding virtual images. And a module allows for high-speed imaging without the need to make significant modifications to a standard microscope.
A range of approaches
Historically, new imaging methods have incorporated the concept of spatial and/or temporal multiplexing wherein some or all image pixels or voxels are illuminated and recorded simultaneously. The underlying rationale is to parallelize and thus speed up image illumination and/or detection. This contrasts with the sequential pixel-by-pixel image acquisition traditionally enabled in confocal laser scanning microscopy (CLSM) or multiphoton microscopy (MPM).
Furthermore, such parallelization (in 1D, 2D, or even 3D) reduces the overall energy load on the imaged sample from illumination and yet maintains a sufficient image signal-to-noise ratio during each image frame acquisition. It maximizes the excitation and collection efficiency in the photon-scarce imaging situations (which are very common in fluorescence microscopy and MPM). But it also minimizes photobleaching and phototoxic effects, and thus preserves biological specimen viability even for prolonged imaging sessions. Over the years, various multiplexing strategies, notably multifoci or multi-light-sheet imaging, have had considerable impact on a range of microscopic methods.
Imaging goes multiplexed
Traditionally, CLSM or MPM relies on the galvo mirror to perform laser scanning. It typically offers a line-scan rate of tens of kilohertz, which allows 2D live bioimaging at a video rate (~10 fps). For example, a 12-kHz resonant galvo mirror module could capture 512 × 512 pixels. The speed of the galvo mirror, however, is fundamentally limited by the mechanical inertia. At the same time, nonmechanical laser scanners, such as acousto-optic and electro-optic deflectors, achieve faster scanning up to hundreds of kilohertz, at the expense of angular scan range and the number of resolvable scan points.
However, these methods still do not meet the growing demand for speed in modern applications. To achieve the kilohertz 2D frame rate in MPM that is required for recording the fast neuronal voltage dynamics in living brains, a line-scan rate well beyond 1 MHz — a speed regime unattainable in any classical laser scanners — is needed.
Several novel strategies have been developed over the past few years to address this limitation. They generally share the same design rationale — to parallelize illumination with multiple scanning foci (for example, using a spinning disk, a microlens array, diffractive optical elements, and a spatial light modulator), a scanning 1D line beam, or 2D/3D wide-field illumination (Figure 1). They also may employ light-sheet imaging, light sculpting by temporal focusing, and light-field imaging, and/or multiplex image acquisition and reconstruction of the image computationally, such as with tomographic methods or compressed sensing. These multiplexing methods augment the imaging speed and/or reduce the energy load to the sample at the expense of different imaging specifications, including compromised spatial resolution (especially at deeper tissue imaging depth) and increased computational overhead and complexity of image reconstruction.

Figure 1. Common strategies for multiplexing illumination and detection. Discrete array of scanning foci (in 1D, 2D, and 3D) (top). Continuous 1D line beam,
2D light sheet, and 3D volumetric illumination (bottom). In all of these methods, the image signal is detected from above (z-axis). Courtesy of Kevin Tsia/The University of Hong Kong.
Spatiotemporal multiplexing
With the aforementioned infinity mirror approach, by focusing a laser-pulsed beam onto one of a pair of almost parallel plane mirrors (with a typical reflectivity >99.9% and a misaligned angle α <1 milliradian), the beam could be transformed into a set of beamlets. Each beamlet follows a unique multiple (zigzag) light reflection path. The zigzag reflections are progressively denser (or spatially chirped) and are retroreflected along the identical paths, but with different round-trip time delays. In essence, the returning beamlets from the mirror pair manifest themselves from an array of virtual pulsed sources (each has a divergence cone angle the same as α) (Figure 2).
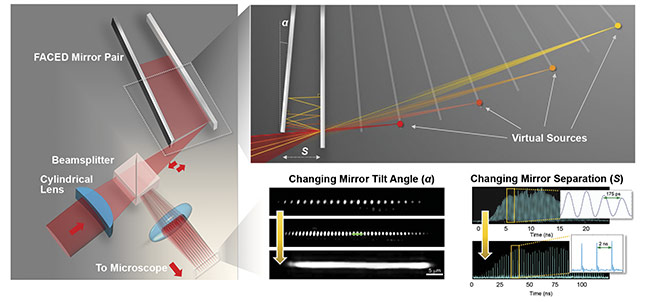
Figure 2. General schematic of a FACED module (left). Understanding virtual source formation in FACED by ray tracing (top right). Reconfigurability of the foci array generated by FACED in terms of spatial profile (by changing the mirror tilt angle) and temporal profile (by changing the mirror separation) (bottom right). Courtesy of Kevin Tsia/The University of Hong Kong.
Positioned between the excitation laser and a microscope, the FACED module can spatiotemporally project the virtual
sources onto the focal plane of a microscope as an ultrafast line-scanning beam — all-optically and passively. FACED bypasses the use of active beam scanners and their speed limitations and achieves a line-scan rate beyond 1 to 10 MHz, governed by the repetition rate of the laser. A high-speed 2D imaging frame rate beyond 1000 fps can readily be achieved when the line-scanning foci are swept by a fast galvo scanner along the orthogonal direction.
The scanning foci generated by FACED are also reconfigurable. The total number of scanning foci (N), and thus the field of view, can flexibly be scaled by adjusting the geometry of the FACED mirror module (such as the mirror separation, misaligned mirror angle, and numerical aperture of the input light cone). For example, N = 80 with neighboring temporal separation of 2 ns; or N > 150, with the temporal separation of 100 ps (Figure 2). The free-space retroflection property of FACED introduces no distortion in the shape of each virtual source and thus ensures diffraction-limited scan spots across the entire virtual source array. Also, the minimal mirror loss (reflectivity >99.5%) in FACED is particularly essential for the scenarios where photon budget is limited, especially multiphoton excitation. Simply based on a pair of plane mirrors, FACED can easily be incorporated into existing microscopes — in various imaging modalities and contrasts, such as bright-field, phase contrast, one-photon fluorescence, and multiphoton fluorescence — to enable high-speed imaging with minimal hardware or software modification.
Further multiplexing in 3D
Based on the same infinity mirror concept, multiplexed imaging can further be extended to 3D, where the entire continuous volume is excited and read out within a volume frame time. This is simply done by combining the infinity mirror with additional relay optics (involving a cylindrical lens) such that virtual sources generated by the mirror pair can be transformed into a dense light-sheet array (>30 sheets). This results in a scanless volumetric excitation, in contrast to the standard light-sheet imaging strategy, in which a single light sheet is scanned across the specimen to obtain the 3D image information (Figure 3).
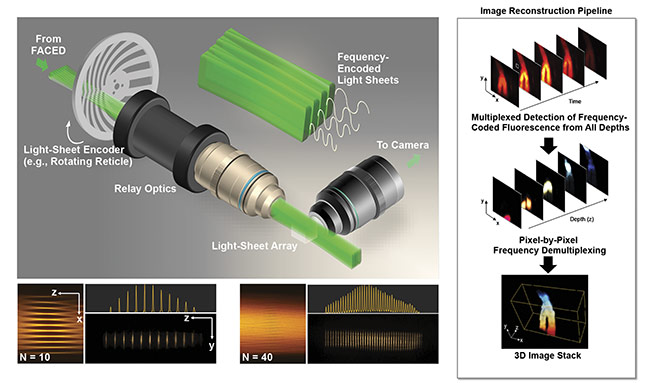
Figure 3. General schematic of a coded light-sheet array microscopy (CLAM) setup (top left). Reconfigurability of the light-sheet array profile generated by CLAM (bottom left). Imaging reconstruction pipeline in CLAM (right). N: number of light sheets. Courtesy of Kevin Tsia/The University of Hong Kong.
Recent advancements in light-sheet imaging have also demonstrated similar parallelization by multi-light-sheet illumination. These methods — most of which are limited to sparsely sampling the imaged volume with a handful of light sheets to reduce the illumination artifacts due to the coherent light scattering (for example, speckle noise) in tissue — commonly resort to coherent beam interference, beamsplitting, or wavefront shaping.
Light-sheet array generation by infinity mirror can flexibly be reconfigurable in the density of light sheets, and, more importantly, the coherency between adjacent light sheets. This is because the path length difference between virtual sources can be tuned across several orders of magnitude in free space (from millimeters to tens of meters). Hence, incoherent superposition of densely packed light sheets can be achieved when the path length difference is adjusted to be larger than the coherence length of the laser source. This attribute is essential in minimizing the illumination artifact and speckle effect, especially in the deep-tissue imaging scenarios.
In this method, a continuous-wave laser, instead of a pulsed laser, can be adopted for parallelized optically sectioned 3D image capture. Apart from the infinity mirror, two additional “tricks” are needed: multiplexed light-sheet coding and extended depth of field (eDOF). First, each continuous-wave light sheet is intensity modulated with a unique temporal code. Therefore, the fluorescence emission signal from a particular section (for example, the kth plane) along the depth within the specimen is tagged with the temporal code corresponding to the kth coded light sheet.
By using the signal multiplexing concept practiced in communication networks (for example, code or orthogonal frequency division multiplexing), it becomes possible to retrieve the image information at each 2D plane, given that the orthogonality of the codes is achieved to effectively offer optical sectioning. To maximize the image depth coverage, the concept of eDOF, or obtaining in-focus images across all the planes simultaneously, could be implemented. In this way, sequential axial scanning (scanning the detection objective lens) is not necessary for volumetric imaging.
A variety of eDOF methods are compatible with light-sheet imaging. They mainly involve point spread function engineering (for example, through nondiffracting beam generation or simply by harnessing spherical aberration). This method, which exploits fully parallelized 3D illumination and detection, is called coded light-sheet array microscopy (CLAM) (Figure 3). Not only can it mitigate the speed limitation imposed by the common serial image acquisition (pixel by pixel, line by line, or plane by plane) and thus allow fast volumetric rate (video or beyond), but it also further minimizes photobleaching and photodamage by avoiding repeated excitation of out-of-focus fluorescence within the volume.
Enabling new applications
The expansion of multiplexed imaging is expected to enable a wide range of applications, from high-speed and long-term dynamical monitoring, high-throughput imaging cytometry to large-scale image-based screening (for example, histopathology). Following are a few examples in the context of the infinity-mirror-based imaging approaches (such as FACED and CLAM).
Kilohertz multiphoton imaging
It remains a critical yet daunting task in neuroscience research to monitor neural signaling at high spatial (synaptic and cellular) resolution and temporal (millisecond) resolution. This is the key to visually dissecting the intricate mechanisms of neural activity in the intact brains of behaving animals. Particularly challenging is to image membrane voltage changes, the most direct measure of neuronal activity, due to their millisecond dynamics. Two-
photon fluorescence microscopy (2PFM) stands out among the existing optical brain-imaging methods because it offers submicron spatial resolution even in the presence of strong light scattering in brain tissue2. However, the imaging speed of the current 2PFM approaches could not cope with the speed required to record neuronal (voltage) dynamics at millisecond resolution in a living brain.
Other high-speed 2PFM methods (up to 1000 fps) have been developed (for example, random-access 2PFM using acousto-optic deflectors, multifoci generation and scanning by microlens array, and tomographic methods) at the expense of imaging field of view, spatial resolution, out-of-focus fluorescence background, and optical system or computational complexity. Enabling a full-frame 2D imaging rate beyond 1000 fps, FACED 2PFM represents a promising candidate for spatiotemporal monitoring of calcium activity, transmitter release, and membrane voltage at kilohertz rate in vivo. Notably, combined with the use of a state-of-the-art genetically encoded voltage indicator — the soma-targeted ASAP3 (accelerated sensor of action potentials) — FACED 2PFM made the first visualization of both the spikes and subthreshold electrical events from neuronal populations deep in the brain in head-fixed awake mice (Figure 4)3.

Figure 4. Applications of multiplexed imaging enabled by FACED. Multiphoton brain imaging at a kilohertz frame rate (top). High-throughput imaging flow cytometry and single-cell image-based analysis (middle). 3D light-sheet imaging for gentle dynamical monitoring and high-throughput 3D histopathology
(bottom). EpCAM: epithelial cell adhesion molecule; t-SNE: t-distributed stochastic neighbor embedding; NS: nutrient starvation. Courtesy of Kevin Tsia/The University of Hong Kong.
High-throughput imaging cytometry
The ultrafast line-scanning operation in FACED also makes the technique ideal for high-throughput imaging cytometry in the on-the-fly configuration, particularly when imaging cells in fast microfluidic flow. Because of its compatibility with other imaging modalities, FACED imaging cytometry could represent a powerful tool for exploiting and integrating all the information-rich morphological content of individual cells at the throughput and content that are not achievable or affordable with current cell-based imaging technologies. Such morphological information includes the biophysical and mechanical properties given by bright-field image contrast and the biochemical signatures provided by fluorescence image contrast.
With advanced computer vision — especially when assisted by deep learning — this technology could be a powerful single-cell analysis tool that enables image-based cell profiling for disease diagnosis (such as identifying disease-specific phenotypes of cells) and drug discovery process (such as screening for mechanisms of action of drugs)4-5. For example, a FACED imaging flow cytometer has been developed for investigating the subcellular morphological changes of the lysosome-labeled cells in both the starvation-induced and drug-induced cell death at an imaging through-put of 75,000 cells/s6 (Figure 4). A similar FACED imaging flow cytometer — with both bright-field and fluorescence image contrasts — has also been employed to classify and analyze the cancer cell population spiked in a heterogeneous human blood sample7.
Volumetric light-sheet imaging
CLAM enables full 3D parallelization in illumination and detection, and thus results in lower photodamage and phototoxicity, compared with the standard CLSM and MPM. Hence, it could be particularly well suited for developmental biology research for which long-term dynamical volumetric live imaging of cells and organisms is essential. In this context, it becomes increasingly important to track individual cells along various cellular pathways (for example, when studying morphogenesis) over several hours or even days across an entire embryo (such as zebra fish, Drosophila, or mouse).
Another advantage of CLAM is its 3D imaging speed. While the first demonstration of CLAM showed a 3D imaging rate of 10 vol/s, it has no fundamental limitation in scaling to a higher volume rate as sCMOS camera technology continually advances (for example, to >10,000 fps in the state-of-the-art sCMOS camera). Apart from high-speed dynamical imaging of living specimens, CLAM could also find new impact in high-throughput 3D histopathological investigation of large, archival biological samples (such as a whole brain).
This is particularly appealing when combined with the rapid development of chemical tissue-clearing methods that enable high-resolution thick-tissue imaging by homogenizing the refractive index throughout the specimen without disrupting its anatomical structure through chemical treatments. CLAM was applied to image tubular epithelial structures and the glomeruli in the optically cleared mouse kidney, as well as to the blood vasculature in the mouse intestine. All were visualized at the volume rate of ~6.4 vol/s (using a total of up to 34 multiplexed light sheets without any beam scanning or objective actuation) (Figure 4).
It is anticipated that CLAM, combined with automated real-time 3D image stitching, could significantly accelerate the workflow in ultralarge-scale 3D whole-organ imaging and, therefore, in comprehensive anatomical investigations.
For a long time, development of microscopy has focused on improving the spatial resolution in life science research, leading to a wide spectrum of superresolution imaging tools. Increasing imaging speed has not been a priority in microscope design criteria until very recently.
With the application of technologies such as FACED and CLAM, and techniques to preserve or augment image quality (for example, adaptive optics and deep learning-assisted computer vision), imaging speed/throughput can now be a priority specification in modern microscope design and development. Emergent concepts of multiplexed imaging will continue to be fueled by the increasing need for high-throughput imaging of single cells, tissues, organoids, and organisms, and the high-speed monitoring of biological dynamics, notably neural activities.
Meet the authors
Jianglai Wu, Ph.D., received his doctorate in physics from Hong Kong Baptist University. He worked as a postdoctoral fellow at The University of Hong Kong and the University of California, Berkeley from 2015 to 2020 before becoming a principal investigator at the Chinese Institute for Brain Research in Beijing. His research focuses on developing novel techniques for brain imaging; email: [email protected].
Na Ji, Ph.D., received her Bachelor of Science in chemical physics from the University of Science and Technology of China. She received her doctorate in chemistry from the University of California, Berkeley. She started working as a postdoctoral fellow at Janelia Research Campus of the Howard Hughes Medical Institute in 2006, before becoming a group leader there. She returned to UC Berkeley in 2016 and is currently an associate professor in the Department of Physics and the Department of Molecular and Cell Biology; email: [email protected].
Kevin K. Tsia, Ph.D., received his doctorate in the electrical engineering department at the University of California, Los Angeles. He is a professor in the Department of Electrical and Electronic Engineering and the program director of the Biomedical Engineering Program at The University of Hong Kong. His research interests cover a broad range of subject matters, including ultrafast optical imaging for imaging flow cytometry and cell-based assay high-speed in vivo brain imaging computational approaches for single-cell analysis; email: [email protected].
References
1. J. Wu et al. (2017). Ultrafast laser-scanning time-stretch imaging at visible wavelengths. Light: Sci Appl, Vol. 6, p. e16196.
2. N. Ji et al. (2016). Technologies for imaging neural activity in large volumes. Nat Neurosci, Vol. 19, pp. 1154-1164.
3. J. Wu et al. (2020). Kilohertz two-photon fluorescence microscopy imaging of neural activity in vivo. Nat Methods, Vol. 17, pp. 287-290, www.doi.org/10.1038/s41592-020-0762-7.
4. K. Lee et al. (2019). Quantitative phase imaging flow cytometry for ultra-large-scale single-cell biophysical phenotyping. Cytometry A, Vol. 95A, pp. 510-520,
www.doi.org/10.1002/cyto.a.23765.
5. J. Caicedo et al. (2017). Data-analysis strategies for image-based cell profiling. Nat Methods, Vol. 14, pp. 849-863, www.doi.org/10.1038/nmeth.4397.
6. W. Yan et al. (2018). A high-throughput all-optical laser-scanning imaging flow cytometer with biomolecular specificity and subcellular resolution. J Biophotonics,
Vol. 11, Issue 2, p. e201700178.
7. J. Wu et al. (2017). Multi-MHz laser-scanning single-cell fluorescence microscopy by spatiotemporally encoded virtual source array. Biomed Opt Express, Vol. 8, pp. 4160-4171.