Darryl McCoy and Marco Arrigoni, Coherent, Inc.
The first article in this two-part series showed how the drive for higher spatial resolution is dominating developments in CW lasers for visible microscopy in the life sciences. In part two, we will see how the need for faster images at greater depths in living tissue is the dominant driver in lasers for multiphoton microscopy, in large part because of dynamic developments in the well-funded field of neuroscience.
Neuroscience has become the key application for multiphoton microscopy because of its multi-billion-dollar funding level and the technical requirement to provide neuronal images in the cortex of living animals. Besides the fundamental drive to better understand how the brain processes information, from the synaptic to the network level, increased life expectancy brings an emphasis on improved quality of life in old age and a need for a better understanding of how to reduce and treat age-related mental impairments such as Alzheimer’s and Parkinson’s.
This funding is supporting advances across the entire neuroscience field; e.g., new insights at the molecular and cellular levels are being correlated with function at the neural network and actual patient levels. Multiphoton imaging is playing a key role in many of these breakthroughs – and this, in turn, is driving product development in ultrafast lasers.
New tools for imaging
The overarching trends in multiphoton microscopy are faster and deeper in vivo imaging. At the same time, novel neuroscience applications are being enabled by a host of new fluorescent tools and methods, many of which use gene expression in laboratory animals, from insects to mice. The field of optogenetics – pioneered by Dr. Karl Deisseroth and colleagues at Stanford University in California – has enabled an important transition, where light is used not just to image structures, but also to stimulate specific cells, thanks to a class of transgenic proteins: opsins.1
These proteins reside in the cell membrane and act as single-element ion gates that can be controlled by pulses of light. When irradiated at the appropriate wavelength, they enable Ca2+ or other ions to flow across the membrane, leading to a spike in the membrane potential that mimics the normal action potential used for neuron conduction and dendritic signaling processes in neural cell networks. Most optogenetics research has been done so far using simple LED light sources, but an appropriate ultrafast laser can be used for photoactivation of particular sites, possibly in conjunction with a spatial light modulator. A second beam is usually then deployed for imaging the Ca2+ activity using, for example, GECI (genetically encoded calcium indicators) expression.
GECIs represent another important class of relatively new probes. These transgenic proteins fluoresce only in the presence of an increased Ca2+ activity, typically providing higher signal-to-noise than traditional calcium indicators that must be injected in the sample and often making long-term in vivo monitoring a difficult proposition. Plus, by clever insertion of promoter genes, it is now possible to express the GECI in a specific class of neuron, rather than in all the cells in the microscope’s field of view. Commonly used GECIs are optimally excited by two-photon absorption around 930 to 940 nm, although more recent ones also can be excited at wavelengths beyond 1 µm. Because Ca2+ concentration events such as neuron action potential spikes happen on the millisecond timescale, this is another driver for faster imaging, requiring more intense excitation of the sample. And, of course, researchers want to probe how these events ripple through networks, looking deeper into the tissue (i.e., the cortex) than ever before.
Fluorescent proteins such as eGFP (enhanced green fluorescent protein) or YFP (yellow fluorescent protein) were used well before optogenetics tools and GECIs, but in this established arena, there are also several new improvements. One of the most visually spectacular brain imaging methods, pioneered by Lichtman and Sanes, goes by the name of “brainbow.”2 Here, subject animals are transfected with genes for three or more different fluorescent proteins, whose emission peaks at red, green and blue wavelengths. The various cell types in the brain express these proteins in unique stochastic ratios, each characterized by a different ratio of the fluorophores and, therefore, a unique emission spectrum and color signal.
Researchers have shown recently that deep brainbow images can be obtained using two-photon excitation at suitable wavelengths (Figure 1). By splitting a Ti:sapphire (Ti:S) laser beam to excite the sample and to generate a second wavelength by pumping an optical parametric oscillator (OPO), Emmanuel Beaurepaire et al have shown that just two wavelengths can excite all the brainbow expressions by using two photons from the Ti:S laser, two from the OPO and one photon each from the OPO and the Ti:S.3 For this to happen, the arrival of the OPO and Ti:S laser photons on the samples must be synchronized.
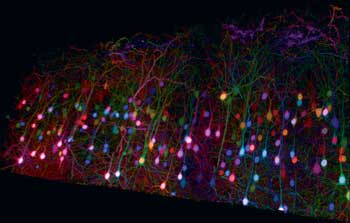
Figure 1. New multiphoton imaging techniques such as “brainbow” are driving continuous improvements to ultrafast laser technology. Here, a trichromatic two-photon imaging of brainbow mouse cortical tissue using wavelength mixing. Courtesy of E. Beaurepaire, P. Mahou, K. Loulier and J. Livet of École Polytechnique, Palaiseau/Vision Institute, Paris (Mahou et al, Nat Meth 2012).
The addition of multiple synchronous wavelengths through the OPO now enables the full potential of multimodal microscopy; e.g., a red fluorescent protein such as TdTomato or mCherry can be used to image certain structural elements with the OPO, while the Ti:S laser output is used to excite some conventional dye. Or, the Ti:S laser can be used to excite eGFP, while the OPO images lipids or cell boundaries via third-harmonic generation (THG) microscopy.
The drive for higher-speed imaging
The transition from static or even video-rate imaging to real-time functional studies of neural processes is increasingly enabled by these new genetic expressions as well as by newer groups of dyes and indicators. These allow researchers to look at a bigger picture, to understand how the interrelationship between cells is responsible for extended system function. Here, speed is paramount; key biochemical processes are on the millisecond timescale and the relative timing of cell activities has been shown to be critical to system function. Thus, frame rates of even hundreds of hertz are sometimes required.
The demand for higher imaging speed also is driven by the need to follow various, often widely separated, cells simultaneously in real time. Just as important, researchers are looking deeper into tissue (in some cases obtaining 3-D images at depths in excess of 1 mm in living mouse cortex). This is being enabled by a move to ever-longer excitation wavelengths to minimize scatter, supported by methods and probes optimized for long-wavelength excitation.
Higher-speed imaging requires more excitation in a shorter dwell time per pixel – i.e., a more powerful laser – but it also requires faster scanning modalities than conventional rastering with actively driven galvanometers. Three important developments here merit mention: resonant scanning, spatiotemporal multiplexing and random access multiphoton (RAMP) microscopy, the latter pioneered by Peter Saggau and co-workers.
Conventional multiphoton excitation microscopes are limited in acquisition speed by the galvanometer mirrors, driven by some type of step function with a typical closed-loop dwell time of several microseconds per pixel. In resonant scanning, one or both of the X-Y axes are scanned open-loop with a low-inertia galvanometer freely oscillating like a tuning fork with sweep speeds as high as 10 kHz. The microscope image acquisition is then slaved to this scanner rather than the traditional opposite approach, resulting in tens of frames per second.
Another way to obtain faster images is to simultaneously scan multiple regions of the sample. Several multifocal methods, including the Nipkow spinning disk, have been used for some time to produce multiple focal spots in the X-Y plane. Fluorescence is collected and spatially sorted on a CCD or CMOS camera. A technique demonstrated by Adrian Cheng allows simultaneous imaging at multiple points along the Z-axis and uses a single photodetector to collect all the fluorescence for high signal-to-noise results. This spatiotemporal multiplexing uses a system of beamsplitters and optical delay lines to split the 80-MHz pulsetrain (i.e., one pulse per 12 ns) from a Ti:S laser into four collinear beams with a 3-ns delay between successive beams.4
The beams are set up to have various divergences, so each of the four times is focused at a different Z depth. Fast time-gating of the signal then allows the fluorescence to be sorted into these four sample depths with no drop in scan speed (Figure 2). But, as with resonant scanning, subdividing the laser output between several sampling points necessitates higher power to achieve good signal-to-noise.
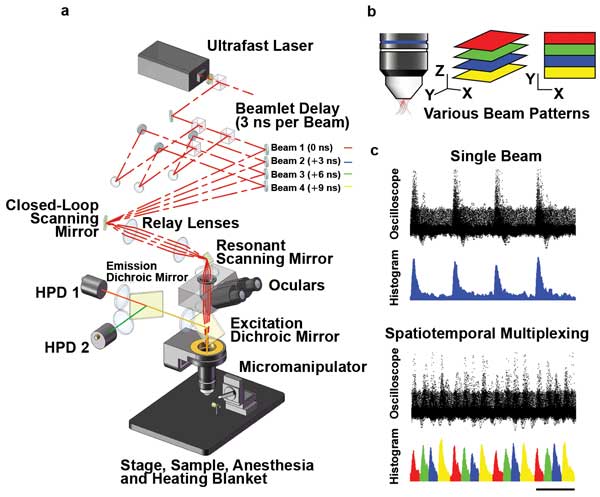
Figure 2. Spatiotemporal multiplexing in multifocal two-photon scanning microscopy: (a) Layout of the prototype microscope. Laser pulses are emitted with 12-ns period from a Ti:S oscillator. The beam is divided into four, delayed by 3 ns each (1 m per 3 ns) and converged on the slow-axis scan mirror aperture, which is then projected onto the objective back aperture. The resulting emitted fluorescence, which is highly scattered, is collected by two hybrid photodetectors (HPDs). (b) Various beam patterns. (c) Comparison of results from a single beam with results from spatiotemporal multiplexing. Courtesy of Portera-Cailliau Laboratory, UCLA.
Maximal flexibility of scan patterns at very high frame rates is achieved when using a pair of acousto-optic deflectors (AODs), as in the RAMP technique (Figure 3). RAMP supports arbitrary access to noncontiguous recording sites, an approach developed to spatially sample strategic sites on neuronal dendrites at very high speed without having to raster-scan the entire dendrite.5 Using diffractive optical elements such as AODs rather than optomechanical beam scanning, Peter Saggau and colleagues have achieved sampling rates of ~50,000/s and, thus, rates as high as 1000 fps for 50 sampling sites. They also extended the approach to three dimensions; without moving the objective lens, 3-D RAMP microscopy enables them to spatially sample sites in a volume at the same sampling rate, using four AODs.6 This approach is now being used to monitor the firing pattern of hundreds of neurons in the intact brains of lab animals.
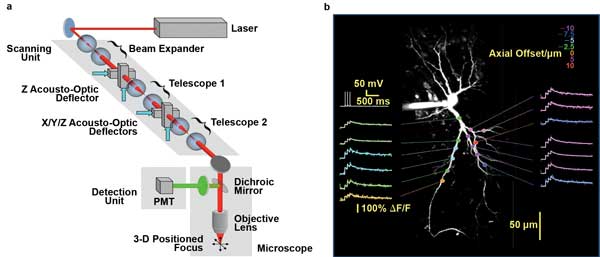
Figure 3. 3-D RAMP microscopy can steer a laser focus within a volume of brain tissue without any moving parts to monitor neuronal activity at very high speed. (a) Scheme of 3-D-RAMP microscope using four AODs. (b) Calcium transients along the dendrite of a single neuron. Recording depth is color-coded. Imaging speed 3000 fps. Courtesy of Peter Saggau, Department of Neuroscience, Baylor College of Medicine.
The use of four AODs adds considerable group velocity dispersion (GVD) and spectral dispersion, which is compensated with yet another AOD. Each AOD transmits only 70 percent of the laser power. The use of all these lossy diffractive optics in series requires a high-power laser with very high beam quality such as the Chameleon Vision from Coherent, the output power of which peaks at more than 3 W.
Better lasers, faster/deeper imaging
Laser manufacturers are responding to these trends in multiphoton imaging by developing turnkey products specifically optimized for the latest methods and experiments. First and foremost, the need for faster and deeper imaging translates directly into a need for higher power and longer wavelengths. The Ti:S gain bandwidth trails off at long wavelengths. Both of these needs have been simultaneously met by increasing the pump power and enhancing the long-wavelength performance of the cavity mirrors. One example is the Chameleon Ultra II, which now provides >5 W of power at the center of the gain bandwidth and usable power out to 1080 nm, currently the longest operating wavelength for any one-box Ti:S ultrafast laser.
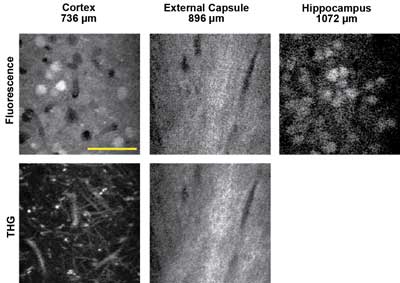
Figure 4. In vivo three-photon excited fluorescence and third-harmonic generation (THG) images of red fluorescent protein-labeled neurons using a 1675-nm excitation source, recorded in three regions of the intact mouse brain. The depth below the surface is indicated for each image. Scale bar = 50 µm. Courtesy of Chris Xu, Cornell University.
Moving to even longer wavelengths (beyond 1.1 µm) now can reduce scattering so effectively that images deeper than 1 mm can be obtained from a living mouse cortex. Of course, this needs both a laser source and suitable probes that can be excited at these wavelengths. The long-wavelength-source requirement is met by combining a compact OPO with the ultrafast laser. This provides turnkey tunable access to the 1000- to 1600-nm region.
There are two excitation approaches for probes in this wavelength region. One is to use red or even infrared-shifted fluorescent proteins (mCherry, TdTomato, IRFP) or certain dyes that can be excited in the 1100- to 1300-nm region. The other is to use three-photon excitation at even longer wavelengths. Chris Xu of Cornell University has been pushing the limit of deep images in several works and recently published images of samples excited at wavelengths as long as 1.6 µm in a three-photon excitation modality. Three-photon absorption is a lower-probability transition than two-photon absorption, so high laser excitation power (or energy) is critical. This and the previous examples illustrate the most recent trends in multiphoton imaging for neuroscience that drive the requirements for higher available power at many excitation wavelengths.
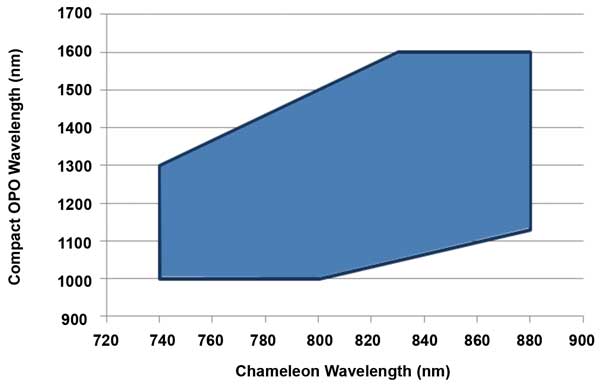
Figure 5. With fan-poled technology, the Ti:S pump and OPO can be independently tuned over a wide range, as indicated in the shaded area of this graph. This independent tuning is a critical advantage for both multiwavelength (e.g., brainbow) and multimodal (e.g., eGFP and THG) imaging applications. Courtesy of Coherent.
Independently tunable OPOs
OPOs were first developed as a means of extending ultrafast laser output to the near-IR (at 1.5 µm and beyond). And there is a growing need to use these longer wavelengths in a variety of multiphoton methods for research in neuroscience. However, growth in the use of multimodal imaging and multiwavelength techniques such as brainbow or simultaneous imaging and optogenetics experiments is creating a demand for turnkey laser systems that can simultaneously produce two wavelengths.
These techniques often can only be fully realized with a laser source that produces two independently tunable wavelengths. This was not possible with traditional nonlinear crystals used in first-generation OPOs, where the tuning of the OPO and of the Ti:S laser could not be separated. This limitation has been removed in the latest OPOs, including the Chameleon Compact OPO, where the use of a fan-poled crystal allows independent tuning of the Ti:S and the OPO (Figure 5).
Meet the authors
Darryl McCoy is the director of product marketing at Coherent Inc.; email: [email protected]. Marco Arrigoni is the technical marketing director at Coherent; [email protected].
References
1. R. Prakash et al (2012). Two-photon optogenetic toolbox for fast inhibition, excitation and bistable modulation. Nat Meth, Vol. 5, Issue 12, p. 1171.
2. J. Livet et al (2007). Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature, Vol. 450, Issue 7166, p. 56.
3. P. Mahou et al (2012). Multicolor two-photon tissue imaging by wavelength mixing. Nat Meth, Vol 9, Issue 8, p. 815.
4. A. Cheng et al (2011). Simultaneous 2-photon calcium imaging at different cortical depths in vivo with spatiotemporal multiplexing. Nat Meth, Vol. 8, Issue 2, p. 139.
5. V. Iyer et al (2006). Fast functional imaging of single neurons using random access multi-photon microscopy (RAMP).
J Neurophys, Vol. 95, Issue 1, p. 535.
6. G.D. Reddy et al (2008). Three-dimensional random access multiphoton microscopy for functional imaging of neuronal activity.
Nat Neurosci, Vol. 11, p. 713.