Digital illumination overcomes the limitations of fluorescence imaging
Dr. Margaret L. Gardel and Dr. Yvonne S. Aratyn, University of Chicago; Dr. Thomas J. Hope, Northwestern University; Robert Nowak and Andrew DeSimone, Photonic Instruments Inc.; and Linda Smith, Peloton Diagnostics Corp.
Live cells represent vital model systems for studying organism development and human disease. Functional, molecular and morphologic quantitative fluorescence imaging techniques are important tools that provide data about the biochemical, genetic or pharmacological processes – noninvasively, in real time and repeatably. The potential of fluorescence imaging of live cells remains untapped because of limitations that include the spatiotemporal-based processes inherent in living cells. Digital illumination, however, in combination with confocal and wide-field microscopy, promises to overcome this in live-cell imaging.
Fluorescence recovery after photobleaching (FRAP) and photoactivation experiments targeting multiple regions of interest (ROI) are employed to identify and analyze the mobility of proteins incorporated within subcellular structures. In a FRAP experiment, structures within the cell are visualized by expression of fluorescent protein fusion constructs. By forcibly bleaching in a targeted region of interest and imaging the recovery of fluorescence intensity, it is possible to extract the unbinding or mobility kinetics of the fluorescent-tagged protein. If the observed protein is mobile, eventually the signals in multiple ROIs will become equal; however, if signals do not reach a point of equilibrium, this suggests that a significant portion of the protein may be immobile. This method enables researchers to probe the stability of various macromolecular assemblies within the cell.
Critical to maintaining data integrity in temporal-based live-cell studies is digital illumination – the ability to simultaneously excite entire and multiple regions of interest in real time with zero delta time. It allows researchers to collect FRAP and photoactivation data without recovery or diffusion gradients. FRAP experiments may include both fast and slow recovery components. Digital illumination can capture the fast component in an area larger than the diffraction limit, whereas the intrinsic delta time of a point scanner can miss or obscure this event.
Spatiotemporal-based studies
Dr. Margaret L. Gardel, assistant professor of physics at the University of Chicago, applied digital illumination to FRAP methods when analyzing both the qualitative and quantitative behavior of the F-actin cytoskeleton as it regulates cell adhesion and migration. Gardel, employing a spinning disk confocal microscope, studied the dynamics and turnover of the F-actin cytoskeleton in adherent cells. In cell migration, the dynamics of the F-actin cytoskeleton must be precisely regulated in space and time to guide changes in cell morphology. Using this technique, she and her team captured these dynamics with high spatiotemporal resolution. They resolved changes in architecture and dynamics that occurred across the cell to understand how variations in kinetics are correlated to physical behaviors and alterations in cell morphology.
15-second sequence
Figures 1a-c show a time-lapse sequence of a photobleaching experiment. FRAP shows U2OS human bone osteosarcoma cells expressing GFP-actin concurrently within several regions. The cell is imaged every 15 s. In reference to the elliptically shaped digitally illuminated photobleached zones, the actin has a half-time recovery of about 30 s. In reference to the circular-shaped digitally illuminated photobleached zone in the cell center, the diffuse cytoplasmic actin simultaneously undergoes rapid recovery.
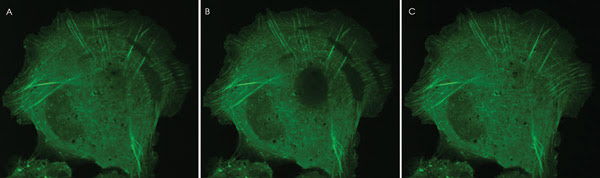
Figure 1. (a) Cell before photobleaching event; (b) cell immediately after photo-bleaching with digital illumination; (c) cell 30 s after photobleaching event.
Photobleached stress fibers show the movement of the bleached zone along the stress fibers over time. The movement of the zone is distinctly in the direction of the cell center, along the stress fibers (tip to base), with a retrograde flow rate of 0.3 μm/min. The photobleached zone also undergoes a recovery in fluorescence of approximately 60 percent of its original intensity, with a half-time of 30 s. Therefore, actin associated with stress fibers undergoes retrograde flow while also exchanging with the soluble pool.
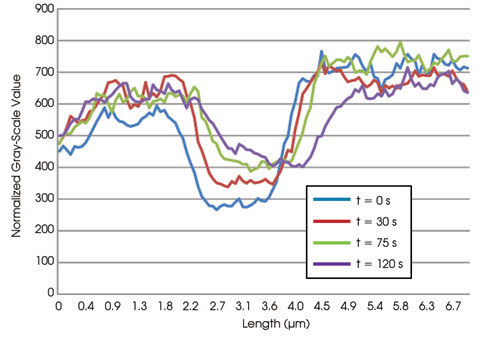
Table 1. Retrograde flow and normalized recovery of fluorescence over time.
Digital illumination allowed Gardel and her team to make observations and to validate cell migration data, which would not have been possible if multiple regions, including diffuse cytoplasmic actin in the cell center, had not been illuminated simultaneously with zero delta time. To measure protein dynamics in a FRAP experiment, image acquisition must be faster than molecular exchange. If this is not achieved, the fluorescence recovery process that gives information about both protein kinetics and time scales for exchange is not resolvable.
Dr. Thomas J. Hope, biology professor at Northwestern University in Chicago, has employed photoactivation and photobleaching experiments while studying the mobility of HIV and its proteins. Critical to his experimental design is the ability to photoactivate simultaneously multiple ROIs within a living cell.
In a typical FRAP experiment, HeLa cells were transfected with an HIV Gag-GFP fusion protein – the HIV protein that forms new viral particles (Figure 2). Images were captured at 5-s intervals, followed by deconvolution.
![Cell-Feat_Fig-2a_gT06[1]linda.jpg](https://www.photonics.com/images/Web/Articles/2008/11/1/Cell_Feat_Fig_2a_gT06_1_linda.jpg)
Figure 2. (a) Cell before exposure to digital illumination, t 5 0 s; (b) digital illumination of two lines with laser, t 5 15 s; (c) resulting region of bleaching, t 5 20 s; (d) photo-bleached region recovering, t 5 35 s.
At t 5 0 s, the expression of the fluorescent-tagged protein in the living cell is at steady-state localization. The protein is either rapidly mobile, anchored or in a combination of the two states, where some protein is moving and some is anchored. After ROIs are digitally illuminated or bleached at t 5 15 s, information about moving and anchored populations can be determined by the way the signal recovers in the bleached region. Anchored protein continues to occupy its position and cannot be replaced by a tagged protein that is not bleached.
The protein in the example is a derivative of Gag that is mostly mobile, and so the fluorescent signal is mostly replaced. Hope showed that, as domains of the Gag protein were deleted, the anchored or immobile fraction was decreased, which correlated with the protein’s ability to form oligomers. Traditional galvo mirror-based laser scanning illumination could not have bleached the regions of interest simultaneously; thus, time-based comparison of fluorescence emission signals would have been impossible.
Phototoxicity
An inherent limitation in fluorescence imaging is achieving sufficient excitation energy while maintaining the integrity of the sample and fluorophore. When every pixel is illuminated evenly, as with conventional confocal microscopy, pixels without fluorophore molecules present are illuminated, compromising the integrity of the sample. Likewise, when pixels with high concentrations of fluorophores are illuminated with higher intensity than necessary, the image saturates with high signal-to-noise ratio, damaging the sample and reducing the dynamic range. With digital illumination, precision in exciting complex sample geometries with zero-delta acquisition time avoids modifications of the sample and inadvertent photobleaching, which minimizes or even eliminates damage.
As an example, phototoxicity was a major concern in Gardel’s utilization of FRAP to study F-actin cytoskeleton dynamics, especially when incorporating a 405-nm laser source. This source was preferred because not only did it feature the desired high excitation efficiency but it also was spectrally far from the experiment’s imaging laser and would not interfere when combined with a single dichroic filter. This meant that, during imaging, it was unnecessary to switch dichroic filters to block the excitation source, so no time delays resulted. And because the digital micromirror device (DMD) featured diffraction-limited precision illumination of complex sample geometries, sample cell integrity was not compromised, and the advantages of a 405-nm excitation source could be exploited.
Autofluorescence
Another inherent limitation in fluorescence imaging is autofluorescence from local nontargeted, but fluorescing, biological compounds. This phenomenon leads to background noise and reduced sensitivity. Simultaneously illuminating complex sample geometries with a DMD device allows for greater precision and minimizes the effects of autofluorescence so that higher sensitivities and improved levels of quantitation can be achieved.
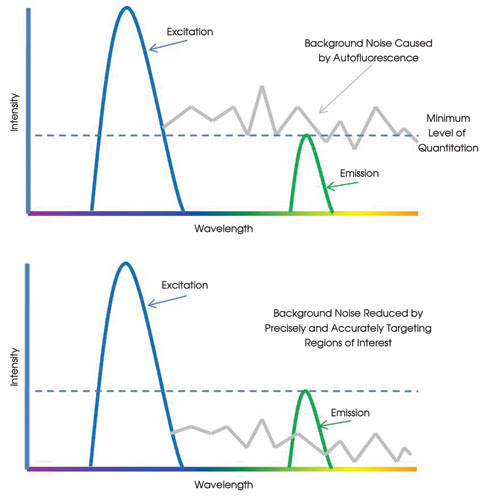
Table 2. Background noise limiting quantitative analysis.
Techniques such as FRAP, photoactivation and photoconversion require the exposure of limited cell areas. Such controlled illumination is available with scanning confocal microscopy. Conventional wide-field fluorescence microscopes are not amenable to techniques requiring illumination of restricted areas during live-cell observation. To address this problem, the typical solution is a point source of laser light that is focused on an area only several microns in diameter. Digital illumination allows the user to configure the shape and size of the area of illumination so precisely and flexibly that specific cellular organelles and structures can be illuminated.
In Hope’s study of the dynamics of intracellular movement of HIV proteins fused to fluorescent proteins, he digitally illuminated the exact cellular regions containing the protein. Controlling the level of illumination minimized background noise and enabled the researchers to gain important insights into how new viral particles are formed in infected cells.
Furthermore, to understand how HIV can penetrate the epithelial barriers of the human genital tract, Hope’s team developed a photoactivation technique enabling the detection of virions in tissue sections. To identify faint fluorescent signals of labeled virions in the presence of the extensive background associated with autofluorescence of tissue sections, the investigators used particles containing photoactivatable GFP.
After defining the background by capturing a Z-series through the tissue section, they photoactivated the sample. The tissue section is imaged again in the green channel, and any new signals identified in the second image acquisition are considered viral particles. Employing digital illumination reduces autofluorescence of surrounding regions and, ultimately, background noise so that quantitation of faint fluorescent signals is achieved.
DMD technology holds the promise of revolutionizing live-cell imaging and is poised to do the same for other fluorescence imaging applications. For the first time, researchers can simultaneously and accurately excite fluorophores in multiple regions of interest with complex geometries and with zero delta time. With this tool, they are realizing unprecedented discoveries in the life sciences.
Meet the authors
Dr. Margaret L. Gardel is an assistant professor of physics and Dr. Yvonne S. Aratyn, a postdoctoral scholar, both at the University of Chicago; Dr. Thomas J. Hope is a biology professor at Northwestern University in Chicago; Linda Smith is the founder of Peloton Diagnostics Corp. in Needham, Mass.; Robert Nowak is the founder of Photonic Instruments Inc. in St. Charles, Ill.; and Andrew DeSimone is responsible for the technical vision at Photonic Instruments; e-mail: gardel_at_uchicago.edu; [email protected]; [email protected]; [email protected]; [email protected]; and [email protected].
How It Works
The core of digital illumination is the digital micromirror device (DMD), a high-speed and highly efficient semiconductor-based “light switch” array of up to 2 million hinge-mounted addressable, tiltable microscopic mirrors. It is a mass-produced spatial light modulator based on mature semiconductor material systems and processing. Numbering more than 8 million in the field, the DMD is also the core of consumer electronics products such as HDTVs and projectors.
Digital micromirror arrays are shown digitally illuminating a sample. When a DMD chip is coordinated with a digital video or graphic signal, a light source and beam delivery optics, its mirrors reflect a digital image onto the sample. Infinitely complex geometries of fluorophore excitation patterns are simultaneously mapped onto the sample. In combination with continuous-wave or arc lamp light fluorescence excitation sources and laser scanning confocal, spinning disk confocal or wide-field microscopes, the computer-controlled DMD spatial light modulator produces a diffraction-limited mask at the specimen plane, at the image detector and within the microscope eyepiece field of view.
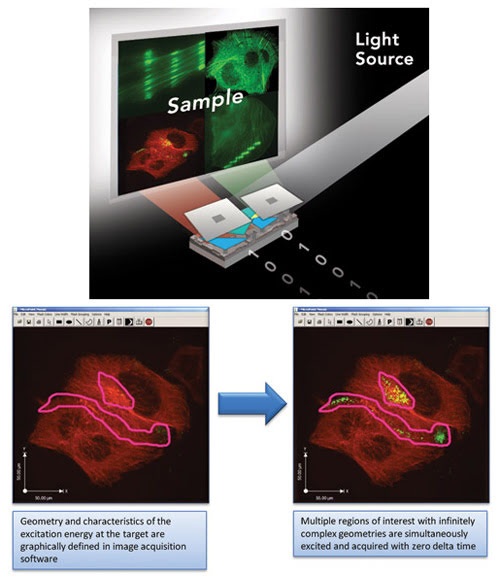 As with traditional galvo mirror scanning systems, DMD technology allows the user to apply pixel-by-pixel illumination where needed and for as long as necessary. However, the inherent advantages of the DMD technology uniquely enable simultaneous pixel-by-pixel illumination of multiple regions of interest and infinitely complex sample geometries with zero delta image acquisition time. There is no scanning of the sample and, thus, no time lapse between illuminating pixels in the mask. This is not possible with traditional galvo mirror technology, which scans the regions of interest over a period of time.
As with traditional galvo mirror scanning systems, DMD technology allows the user to apply pixel-by-pixel illumination where needed and for as long as necessary. However, the inherent advantages of the DMD technology uniquely enable simultaneous pixel-by-pixel illumination of multiple regions of interest and infinitely complex sample geometries with zero delta image acquisition time. There is no scanning of the sample and, thus, no time lapse between illuminating pixels in the mask. This is not possible with traditional galvo mirror technology, which scans the regions of interest over a period of time.
A DMD’s simple planoreflective optical design is readily integrated with the complex optical designs of confocal and wide-field microscopes to realize diffraction-limited imaging with minimal loss over a broad spectral range. Alternative spatial light modulating technologies such as acousto-optic and liquid crystal are less efficient and optically more complex, making them less amenable to diffraction-limited integration with off-the-shelf microscope platforms. This is a critical consideration for maintaining reproducibility of microscope-specific data and for staying within equipment budgets.