Technique used to study protein-protein interactions
Hank Hogan
A team of researchers has paved the way for faster imaging of intracellular events with an advance in microscopy technology. By combining multifocal, multiphoton excitation with time-correlated single-photon-counting detection, the group, from Imperial College London, from the London-based Cancer Research UK and from GlaxoSmithKline, has developed a tool that could be used to study the protein-to-protein interactions that take place when immune cells examine target cells.
The contact between such cells, the immune synapse, has a complex distribution of protein signaling across it. A better understanding of what is going on at the immune synapse would advance biological research and the development of therapeutics.
However, capturing the required information is not easy. “The immune synapse usually does not lie conveniently in the focal plane; it often is necessary to acquire 3-D stacks to permit the immune synapse to be studied,” said Christopher W. Dunsby, a researcher with the photonics group at Imperial College London.
One possible technique is Förster resonance energy transfer (FRET), a process in which an acceptor and donor chromophore interact to change a fluorescent signal. Because FRET happens only when the donor and acceptor are within a few nanometers of each other, the method can serve as a molecular yardstick and provide information on interactions. The FRET signal can be read using fluorescence lifetime imaging microscopy (FLIM). To perform FRET with FLIM readout to study the immune synapse, high-speed three-dimensional lifetime-image stacks of cells must be acquired rapidly. This requirement prompted the development of the new microscopy tool.
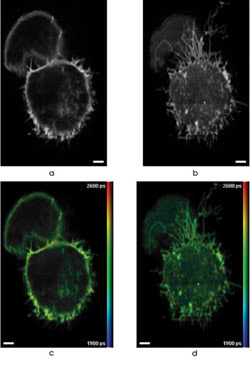
3-D FLIM was performed using multiphoton, multifocal microscopy of cells expressing GFP. Images a and b show fluorescence intensity, and c and d are correspondingintensity-merged false-color of GFP-expressing cells. These are two out of 25 optically sectioned images spaced 2.5 μm apart and recorded with an acquisition time of 60 s per slice. Scale bars are 2.5 μm. Images reprinted with permission of Optics Express.
Multiphoton, multifocal microscopy is well-established because using two or more photons for excitation decreases photobleaching and photodamage out of the focal plane, and using multiple focal points lessens photobleaching and photodamage in the focal plane. The researchers extended the technique by implementing fluorescence lifetime imaging.
They did this with a multielement detector array using a commercially available system from the Berlin-based Becker & Hickl GmbH. This 16-element photomultiplier can perform time-correlated single-photon-counting detection at each element, which allowed photon-efficient detection of the fluorescence decay profile. That is an important capability, given the low levels of fluorescence often associated with live-cell studies.
There were two main challenges in implementing the system. The first was creating the array of excitation spots at the sample. The second was aligning everything so that the fluorescent signal from a given spot was imaged onto the appropriate detector element. The researchers created an array of 16 excitation spots and achieved the proper alignment using a beamsplitter and scanner from LaVision BioTec of Bielefeld, Germany. Using a configuration that did not require the fluorescence signal to be descanned helped the mapping of signals to the sample, Dunsby said.

For a light source, they used a Ti:sapphire Spectra-Physics laser, tuning the output peak as needed from about 700 to 1000 nm. They sent this beam through the beamsplitter and optics so that the foci were spaced about 2.5 μm apart at the sample yet imaged onto separate elements at the detector. They scanned the beams in one direction and moved the stage in the perpendicular direction for a complete image. Fluorescence was captured for a set of 16 lines, and the data was integrated for times that ranged from half a second up to tens of seconds. The integration time depended on the brightness of the sample.
The researchers then processed the data, creating stacks of FLIM information that could be turned into an image or analyzed for quantitative results.
In demonstrations of the system, they looked at fluorescently labeled pollen grains and cells transfected to express green fluorescent protein. They also examined the changes in the fluorescence lifetime of cellular NADH, an important coenzyme found in cells. They examined the autofluorescence from single cells after metabolic inhibition via NaCN, which blocks a process involving NADH within a cell. The work is detailed in the Oct. 1 issue of Optics Express.
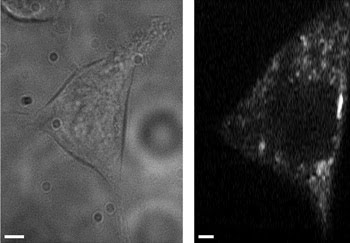
A transmitted light image (left) and an autofluorescence intensity image (right) were averaged over three frames of cellular NADH. Changes in cellular NADH autofluorescence may identify cancerous cells without labeling. These images were taken with a multiphoton, multifocal microscope. Scale bars are 2.5 μm.
These measurements were difficult because of the relatively low fluorescence signal. Simply pumping in more laser power would not increase the signal because of phototoxic effects. Despite such problems, the team revealed a change in the fluorescence lifetime for a small number of cells after stimulation. Work currently is under way to improve upon these results, in part because it has been shown that fluorescence lifetime changes in cellular NADH autofluorescence may provide a way to identify cancerous cells without the need for labels.
Dunsby said that further improvements in speed might be possible through the use of more sensitive detectors or of more foci. Either one or a combination of both would decrease the total amount of time needed to scan a sample. Even with such improvements, however, he doubts whether the current setup could be commercialized successfully.
“The exact configuration that we used requires a combination of beam and stage scanning,” he said. However, there are other possible arrangements that would not suffer from this drawback.