
Method developed for testing and comparing microscope performance
Enables calibration for quantitative microscopy
Gary Boas
Optical microscopes can be found in all areas of biological research:
from studies monitoring translational movement of proteins with fluorescence recovery
after photobleaching to those probing deep into tissue with two-photon excitation
and beyond. In all cases, changes in any number of parameters can result in less-than-optimal
performance. Indeed, performance might differ significantly from microscope to microscope,
in different configurations of the same microscope and even in the same device from
one imaging session to the next.
Edward H. Cho and Stephen J. Lockett of the Imaging
Analysis Laboratory at the National Cancer Institute in Frederick, Md., were keenly
aware of how even slight changes can affect the performance of an optical microscope.
They knew this from their own experiences and because the laboratory is a core facility
with users from throughout the institute. Changes in microscope performance might
come from a smudge on the lens, from dirt left in the microscope, or from normal
wear and tear.
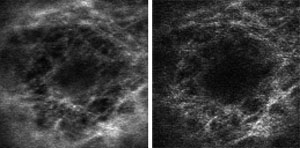
Researchers have developed a means to assess the performance of optical microscopes quantitatively.
Subsequent comparisons of detection efficiency with GFP-tubulin expressing MCF-7
cells yielded a number of interesting findings — indicating, for example,
that high dynamic range can be achieved by reducing the gain (from 950 V in the
left image above to 650 V in the right) and thus the amplification noise. The cell
line was created by Janis Bunker and provided by Mary Ann Jordan and Kathy Kamath.
Specimen preparation and fixation of cells was by M. Katherine Jung.
The investigators searched the literature
for a simple method to assess the performance of a microscope but did not find one
that could provide an absolute measure or that could be used to compare one microscope
with another, “at least not in a convenient way,” Lockett said. So they
developed a methodology, which they described in the July issue of the Journal
of Microscopy.
Assessing detection efficiency
They chose not to evaluate all of the parameters
that factor into a microscope’s performance, but rather focused on the one
they considered to be the most important: the fraction of emitted photons counted
in the image, which they refer to as detection efficiency. The best way to assess
this, they decided, was to place on the stage a light source that emits a defined
number of photons and compare that with the number of photons that contribute to
the image.
They focused on the detection of the
microscope and not on the excitation provided by the light source because, although
the efficiency of the excitation is important, the lasers that come with microscopes
have an enormous excess of power, so any loss there can be offset, Lockett said.
For the method to work, though, they
had to approximate the isotropic emission of fluorescent molecules. They initially
considered placing an extremely small pinhole in front of a conventional light source,
assuming that the light would be diffracted in such a way as to mimic the emission.
Computer simulations showed, however,
that the pinhole would have to be tremendously small and would require an especially
intense source of light. In short, they confirmed that the approach was not a viable
option.
The researchers, therefore, used a
material that diffuses light, eventually choosing a near-lambertian diffuser from
Edmund Optics of Barrington, N.J. The light source they made consisted of this
and an LED emitting at 565 ±30 or 587 nm in a 35-mm glass-bottom dish. Combined
with a mathematical model of the light path for a confocal microscope and detection
electronics, this yielded an excellent approximation of the fluorescence emission.
They used the method to characterize
the performance of two inverted confocal microscopes made by Carl Zeiss Inc. of
Thornwood, N.Y., each outfitted with a 40x, 1.3-NA oil-immersion objective lens,
a dichroic mirror and an emission filter (they varied these to determine how they
affected the efficiency). They acquired images with either a Hamamatsu photomultiplier
tube or the Meta detector that came standard with the microscopes.
They knew the intensity of the light
from the source and, based on the distribution of pixel intensity, determined the
number of photons that contributed to the acquired image. They calculated the efficiency
by dividing the former by the latter, and showed that it fluctuated with the different
mirrors and filters. The relative efficiency was 1.00 with an 80/20 neutral mirror,
1.16 with an HFT458 filter and 0.93 with an HFT458/561 filter. When they switched
to a 63x, 1.4-NA objective, the efficiency was 0.84.
This technique enables comparison of
a microscope’s performance under a number of other configurations and settings,
including pinhole size, detector gain and pixel dwell time. Indeed, experiments
with various settings yielded a range of findings, some confirming existing assumptions,
others providing new insights.
Insights gained
The scientists noted, for example, that the ratio
of the amplification noise to the gain was always 1:4. This told them that increasing
the gain would not produce more efficiency. The other insight is more interesting,
Lockett added.
It is well-known that one can capture
a very large range of intensities within a single image because of the high dynamic
range of the digital image. However, because the achievable dynamic range is drastically
reduced by the amplification noise, it is desirable to decrease this noise. “So
how does one do that?,” he asked. “Well, since the noise is proportional
to the gain, reducing the noise can be achieved by reducing the gain.”
The absolute measurements of detection
efficiency also provide quantitative measurements of biological samples as opposed
to the relative measurements more commonly performed with today’s microscopes.
Thus, they contribute to significant advances in high-throughput imaging, for example,
or in vivo fluorescence imaging of protein dynamics.
All of which is good news for the Lockett
lab, which is generally interested in developing and using quantitative microscopy
to understand both intracellular and intercellular processes.
Currently, the investigators are using
laser scanning confocal microscopy to understand the dynamics and interactions
of proteins in living cells by measuring ensembles of single fluorescently labeled
molecules. This requires detecting the molecules many times over, with the accuracy
of the results increasing with the number of times the measurements are performed.
“Thus, it is vital to operate the microscopes at maximum detection efficiency,”
Lockett explained.
The researchers also are performing
quantitative comparisons of fluorescence from samples imaged in various sessions
and on various microscopes. Such comparisons are possible only when the absolute
detection efficiency of the microscopes is known.
As for the method itself, Lockett noted
that it could be extended to include multiple LEDs in the light source, allowing
measurements at different wavelengths. This would provide for comparison of a microscope’s
performance with different fluorescent dyes, for example.
Published: September 2006