Xin-Hua Hu, East Carolina University and Wavmed Technologies Corp.; Yuanming Fend, Tianjin University; and Jun Qing Lu, East Carolina University
A new method allows fast, label-free cell classification by combining high-contrast image acquisition with automated analysis. This method could open doors for expanded clinical applications such as classification of blood and tumor cells based on their 3-D morphological features.
Biomedical researchers most often carry out rapid assays of large cell populations on a single-cell basis with a flow cytometer. The instrument originated from research using cell counting with laminar flow in the 1950s and from fluorescence measurement in the 1960s. The device can achieve rapid assay with a laminar flow to hydrodynamically focus the core fluid, which contains the cells.1,2 The narrowed core fluid then transports the cells in single file through one or more interrogating beams of incident light.
The optical excitation of a flowing cell leads to two types of light signal emissions. The first type is called a scattered light signal, which has the same wavelength as the incident light and arises from the heterogeneity of the cell's refractive index relative to the surrounding medium. The second type of emission is made up of fluorescent signals from wavelengths that usually are larger than the wavelength of the incident light; they are caused by fluorescent molecules inside the cell. Because most biological cells are not self-fluorescing, cells often must be stained with fluorescent reagents before the flow cytometer can measure them.
Although both types of light signals are acquired from the excited cells for analysis and classification, existing flow cytometers depend mainly on fluorescent signals because the conventional designs for these instruments have limited ability to extract information from scattered light signals.
Existing flow cytometers can be divided into two categories according to their approach to signal acquisition. Most are designed with a single light detector such as a photomultiplier or photodiode to measure signals from both scattered light and fluorescence from stained cells.3 Since both signals are relatively strong in relation to background noise, this design permits signal measurement and acquisition during a time window as short as about 10 µs; this provides a foundation for collecting light signals from flowing cells at rates of up to 104 cells per second, or millions of cells in less than half an hour. At that speed for single-cell analysis, no other instrument even comes close, which is the main reason for its increasing popularity among biomedical researchers.
In 2005, another flow cytometer design began to emerge. Its approach to image acquisition is based on early research dating from the 1980s.4 This system essentially combines a conventional microscope with a flow cytometer to acquire nondiffraction images of cells very quickly. Most flow cytometers, though, rely mainly on fluorescent light, which can provide multiple images per flowing cell, offering additional information on morphology. This benefit comes with a cost, however: lower throughput of 103 or fewer cells per second.
Conventional cytometers: Drawbacks
Despite their wide applications, conventional flow cytometers do have their drawbacks. Fluorescent signals yield important molecular signatures for the interrogated cells, but a single detector can tell only whether —“ and how many — labeling fluorescent molecules exist in a cell. Morphology information is acquired, but the nondiffraction images yield only 2-D projections of 3-D cell structure. More importantly, these nondiffraction images contain complex patterns that are extremely difficult to interpret using automated image analysis with current computer algorithms. Considering that the data stream from an imaging flow cytometer can yield huge amounts of data in a short amount of time, the lack of automated image analysis in real time can present a significant roadblock to taking full advantage of the images for analysis and classification of cells.
Second, measuring fluorescent signals often requires staining cells with multiple reagents, which is a time-consuming process and adds extra costs. In fact, it is widely known that reagent purchase is a major factor associated with the high cost of using flow cytometers. Finally, it is always possible for the fluorescent reagents to interfere with various biochemical processes within the cells under study, and additional tests may be needed to verify and/or minimize this possibility.
Scattered light signals
It has long been known that diffraction imaging methods can be used to acquire 3-D structures of an object under coherent illumination from scattered signals. Soon after the discovery of x-rays more than a century ago, Max von Laue and others found that the patterns of x-ray photons scattered from a crystal sample as recorded by a film can be associated with 3-D lattice structure and constants of the crystal. The observed patterns were used to illustrate the wave nature of x-ray radiation as a result of interference or diffraction among the coherently scattered photons. The results of diffraction research led directly to the birth of x-ray crystallography, which is still widely used by scientists to reconstruct biomolecules in 3-D.
Extending this 3-D image reconstruction method to the optical domain is much more difficult, it turns out, because of the need for highly coherent light sources. Dennis Gabor was probably the first to demonstrate experimentally the feasibility of 3-D reconstruction with optical light.5 However, his significant discovery had to wait for the birth of lasers more than a decade later to be fully appreciated as a high-quality 3-D imaging modality.
As tools for light scattering models have become available, researchers have realized that 3-D reconstruction of an object is possible without a reference beam if a sufficient number of diffraction images are acquired. Nevertheless, 3-D reconstruction of cell structures for rapid assay and classification is difficult, if not impossible, in a flow cytometer setting because multiple diffraction images are needed, and time-consuming inverse calculations must be performed.
Over the past 10 years, we have conducted research on modeling and have performed experimental studies on light scattering by biological cells.6-8 The results have led to the recent development of a new diffraction imaging approach using flow cytometers.9-11 By using confocal fluorescence microscopy and staining the nucleus and mitochondria with various reagents, we made realistic 3-D reconstructions of cell structures. The technique allowed us to import precisely sized structures into a computer model for accurate simulations of light scattering by single cells. Figures 1a and 1b present 3-D cell structures viewed from two perspectives. By assigning different refractive index values to the intracellular components and solving Maxwell's equation, we can obtain the angular distribution of scattered light fields from a cell. With this numerical modeling tool, diffraction images can be simulated by projecting the scattered light intensity on a plane representing an imaging sensor.
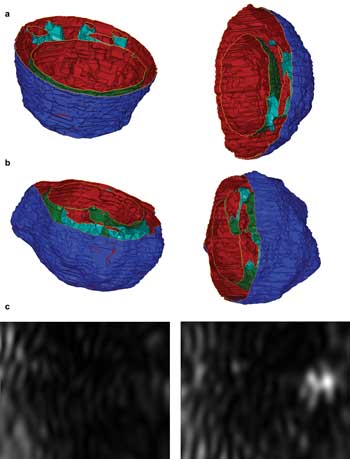
Figure 1. Two views of reconstructed structures of (a) a Jurkat cell derived from a T lymphocyte and (b) an NALM-6 cell derived from a B lymphocyte. The dark-blue surfaces are cell membranes, the light-blue surfaces are aggregated mitochondria, and the green are nuclear membranes. (c) Two simulated diffraction images of side scatters are shown from the same cell structure: The left corresponds to a cell membrane surface with the same index of refraction as that of cytoplasm at nm = nc = 1.35; the right corresponds to the same structure, but its cell membrane surface index is set at nm = 1.50. Courtesy of the authors.
Using different 3-D distributions of intracellular refractive index, diffraction images can be simulated that yield insight into the correlation between diffraction image patterns and 3-D morphological and index distribution features of illuminated cells. Two examples of simulated images, shown in Figure 1c, demonstrate the effect of intracellular distribution of refractive index. Taken together, these results motivated us to incorporate the diffraction imaging into flow cytometry through a new approach — an alternative to reconstruction — in which the image data are analyzed in real time as the "fingerprints" of 3-D optical structures for rapid assay of cells. We have also performed goniometric measurement of scattered light signals from cell suspension samples and have found that polarization-based measurement can provide additional handles for cell classifications, as expected from modeling results.
Designing a new cytometer
Armed with these new insights, we set out to investigate various designs for the flow nozzle and flow chamber for the diffraction imaging flow cytometer we were developing. These elements play a key role in forming a hydrodynamically focused laminar flow — positioning cells accurately at the optical focus of the interrogating laser beam without any index-mismatched interfaces close to the excited cell and at the desired speed.
Several groups have published various designs for flow chambers that acquire diffraction images from cells; these include a glass flow cell with a square, narrow channel and a waveguide-based beam-in-flow design.12,13 None of these fit our requirements for acquiring high-contrast diffraction images with ease of alignment.
After numerous tests and design improvements, we finally arrived at a jet-in-fluid flow chamber design to meet the above requirements. It incorporates a nozzle built in-house to guide the sheath and core fluids into a water-filled cuvette with its sides several millimeters away from the core fluid. The two fluids from the nozzle form a laminar flow in which the core fluid narrows to an exit tube in the cuvette. A 3- to 5-mm-long gap space exists between the extending tip of the core channel in the nozzle and the exit tube to allow efficient collection of scattered light from the biological cell by the CCD camera, without any noise-producing index-mismatched interfaces within the field of view. With the incident laser beam focused at the core fluid just above the exit tube, the positioning of the flowing particles at the laser beam's waist can be accurately achieved in the gap space with no heterogeneity of refractive index near the excited cell. Figure 2 exhibits a close-up view of the nozzle and chamber.

Figure 2. The flow chamber is based on a jet-in-fluid design concept that uses the objective to collect side scatters for diffraction imaging.
After the initial development stage, we constructed two experimental prototypes of a diffraction imaging flow cytometer for conducting cell measurements at our labs at East Carolina University and Tianjin University. The current design allows for the acquisition of two polarization-resolved images per excited cell using a polarizing beamsplitter behind the infinity-corrected microscope objective, which collects side light scatters. Exploiting polarization information from diffraction image data can provide additional parameters for cell analysis and classification, and data from various polarization states of the incident laser beam can be incorporated. By adopting the gray-level co-occurrence matrix algorithm widely used for analyzing human fingerprints, we can extract multiple parameters at a rate of 70 images per second using a 2-GHz laptop computer for cell classification.
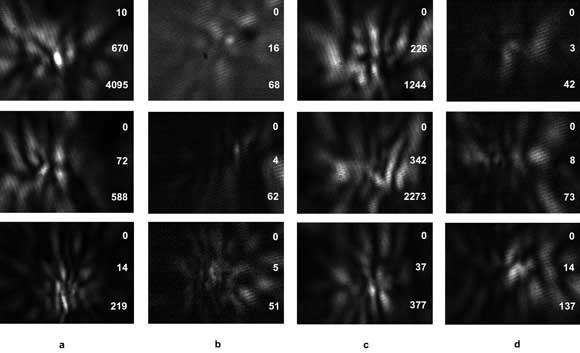
Figure 3. Normalized diffraction images acquired from (a, b) Jurkat cells derived from T lymphocytes and (c, d) Ramos cells derived from B lymphocytes using two 12-bit CCD cameras with an excitation wavelength of 532 nm for each flowing cell: (a) and (c) are images of side scatters with vertical polarization; (b) and (d) are images of side scatters with horizontal polarization. The polarization direction of the incident light beam is vertical (top row), 45° from vertical (middle row), and horizontal (bottom row). The three numbers on the right side of each image are the minimum, average and maximum pixel readings of the 12-bit images before normalization.
With further optimization of the automated image analysis code and parallel execution on multiple GPUs, we expect that, in the near future, real-time diffraction image processing for cell classification can be scaled up to a rate of 103 images per second. Figure 3 shows some examples of recent diffraction image data collected from cultured cells derived from B and T lymphocytes from cancer patients. Quick visual examination indicates that differences in pattern and intensity may be used to perform label-free classifications of these two lymphocyte-derived cell types. A detailed study to test this hypothesis using the gray-level-co-occurrence matrix algorithm is in progress.
Meet the authors
Dr. Xin-Hua Hu is a physics professor at East Carolina University in Greenville, N.C., and chief scientist at WavMed Technologies Corp. in Tianjin, China; e-mail: [email protected].
Dr. Yuanming Feng is a professor and chairman of the biomedical engineering department at Tianjin University in China; e-mail: [email protected]. Dr. Jun Qing Lu is an associate professor of physics at East Carolina University; e-mail: [email protected].
References
1. P.J. Crosland-Taylor (January 1953). A device for counting small particles suspended in a fluid through a tube. Nature, pp. 37-38.
2. W. Dittrich, W. Gohde (March 1969). Impulsfluorometrie bei Einzelezellen in Suspensionen. Z Naturforsch B, pp. 360-361.
3. M.R. Melamed et al (1990). Flow Cytometry and Sorting. 2nd ed. Wiley-Liss.
4. S.H. Ong et al (October 1987). Development of an imaging flow cytometer. Anal Quant Cytol Histol, pp. 375-382.
5. D. Gabor (1948). A new microscopic principle. Nature, Vol. 161, pp. 777-778.
6. J.Q. Lu et al (April 4, 2005). Simulations of light scattering from a biconcave red blood cell using the FDTD method. J Biomed Opt, 024022.
7. R.S. Brock et al (November 2006). Effect of detailed cell structure on light scattering distribution: FDTD study of a B-cell with 3D structure constructed from confocal images. J Quant Spectrosc Radiat Transfer, pp. 25-36.
8. H. Ding et al (2007). Angle-resolved Mueller matrix study of light scattering by B-cells at three wavelengths of 442, 633, and 850 nm. J Biomed Opt, Vol. 12, 034032.
9. K.M. Jacobs et al (2009). Development of a diffraction imaging flow cytometer. Opt Lett, Vol. 34, pp. 2985-2987.
10. K.M. Jacobs et al (2009). Diffraction imaging of spheres and melanoma cells with a microscope objective. J Biophotonics, Vol. 2, pp. 521-527.
11. K. Dong et al (2011). Study of cell classification with a diffraction imaging flow cytometer method. SPIE Proc, 7902.
12. J. Neukammer et al (2003). Angular distribution of light scattered by single biological cells and oriented particle agglomerates. Appl Opt, Vol. 42, pp. 6388-6397.
13. K. Singh et al (February 2004). Analysis of cellular structure by light scattering measurements in a new cytometer design based on a liquid-core waveguide. IEE Proc Nanobiotechnol, Vol. 151, pp. 10-16.