Don’t put off thinking about regulatory approval for a medical device that you are bringing to market – experts say you’ll save big in the long run.
Medical devices must pass strict regulations before they are released for clinical use, and policies vary depending upon where in the world the device is to be marketed. In some countries, navigating the system is so tricky that some companies are beginning to fight back. But wherever you are, the advice is always the same: Start thinking about compliance early.
Unfortunately, many of the biggest medical device markets – including the US, Japan, Brazil, China and Mexico – have the most complex regulations. In Europe, Canada and Australia, things are more straightforward.
All medical devices sold within the European Union (EU) must be CE-marked (“CE” stands for the French phrase “Conformité Européenne,” or “European Conformity”). Depending on the nature of
the device, manufacturers will either be able to self-certify, meaning that they can complete the process themselves and register with the MHRA (Medicines and Healthcare products Regulatory Agency) in the UK, or, in the case of all but the simplest devices, they will need to work with a specialist authority, known as a notified body.
“The process of bringing a medical device to market varies, depending on the type of product you are developing,” said Daniel Jones, director of communications at the UK’s Association of British Health-care Industries. “There are thought to be over 500,000 different products in 10,000 categories.”

FDA Laboratory Building 62 (Engineering and Physics) houses the Center for Devices and Radiological Health.
In Europe, products are classified into the following groups: Class I includes low-risk products and is separated into those products that are not provided sterile or that do not have a measuring function. These are the only devices that can be self-certified. Another category of Class I products includes those that are provided sterile or that do have a measuring function. Medium-risk products fall into Class IIa. Higher-risk products are considered Class IIb or Class III.
“Class I products such as walking sticks, surgical gowns – [things that] represent low risk – are based on a system of self-declaration, where a design dossier is submitted to a Notified Body who then issue[s] a CE mark,” Jones said. “As you move up through the classifications into products that are more complicated, the requirements for evidence become more and more stringent.”
Class III products represent more complicated devices such as surgical implants. For this classification, the system will require clinical trial data, be it new evidence or through the equivalence method, and regulators will carry out inspections of manufacturing facilities.
Although the CE marking process is relatively straightforward, in reality, companies seeking approval for the first time are still advised to seek expert advice from a regulatory specialist. There are many consultancy firms as well as a number of experts who offer guidance through the process.
One such specialist in CE marking and product safety is Conformance Ltd. of Great Hucklow, Buxton, England. Nick Williams, director and senior consultant, said his top pieces of advice are to start thinking about CE marking as soon as possible, to be methodical and to keep good records.
“Ideally, a company would start thinking about the CE marking requirements at the design stage – they can then design the product to meet the requirements,” Williams said. “To CE-mark the product, they would need to obtain a copy of the [Medical Devices] Directive, which contains the legal requirements, and [reference] any associated standards which contain the specifications for design.”
Chris Schorre, vice president of global marketing at the global consultancy firm Emergo Group, agrees with Williams. “If manufacturing a higher-risk product, regulatory compliance must start at the very beginning of the design process,”
he said. “For instance, if you were to design a new Class III device, the MHRA or other government authorities will want to know how you controlled the design of the product, including changes made to it, right from the beginning. With lower-risk products, this is not as much of an issue.”
Fortunately, the process of device approval is harmonized across Europe, which means that rules governing how you bring a product to market are the same in the UK as in any other member of the EU.
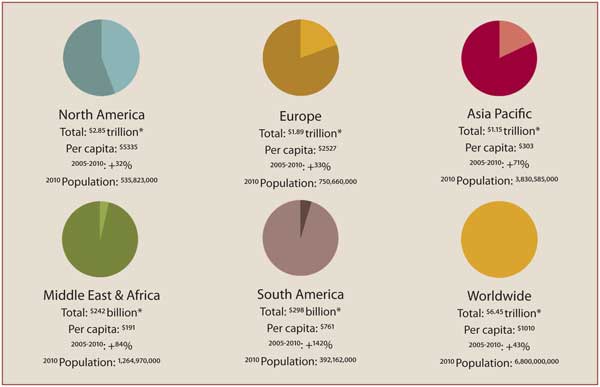
In the US, the process is different. Although the US accounts for 40 percent of global health care spending, it also has fierce competition in all categories, so Schorre’s advice is not to enter a market just because it’s big.
“Look for smaller markets that have good potential for your products but are underserved,” he said. “Also, make sure you understand who pays for your device in that market – private health care insurance only versus government reimbursement.”
Success in the US
The FDA’s Center for Devices and Radiological Health (CDRH) is responsible for regulating firms that manufacture, repackage, re-label and/or import medical devices for sale within the US. In addition, CDRH regulates radiation-emitting electronic products (medical and nonmedical) such as lasers, x-ray systems, ultrasound equipment, microwave ovens and color televisions.
In November 2010, a damning report emerged, blaming unpredictable, inefficient and expensive regulatory processes for jeopardizing America’s leadership position in medical technology innovation.
The report surveyed 204 medical technology companies and found that 44 percent of participants indicated that partway through the premarket regulatory process they experienced untimely changes in key personnel, including the lead reviewer and/or branch chief responsible for the product’s evaluation. A total of 34 percent of respondents also reported that appropriate FDA staff and/or physician advisers to the FDA were not present at key meetings between the FDA and the company.

A physical science technician inspects medical gloves at an FDA laboratory in Irvine, Calif.
The report concluded that factors such as these make the US premarket regulatory process inefficient and resource-intensive, and contribute to significant delays in navigating FDA regulatory processes.
On Jan. 22, 2013, DuVal & Associates of Minneapolis filed a citizen petition on behalf of the Minnesota Medical Device Alliance regarding changes made to FDA regulations in 2009. Although the FDA insists that the changes it made make the process “more reasonable, transparent and predictable,” the petition claims that now, in reality, the “FDA is predictably unreasonable and very transparent about it.”
As in Europe, medical devices in the US are similarly classified into Class I, II and III, with Class III being subject to the highest regulatory control. In general, there are two pathways to approval: 510(k) and premarket approval (PMA).
“510(k) is a premarketing submission made to FDA to demonstrate that the device to be marketed is as safe and effective – that is, substantially equivalent (SE) to a legally marketed device that is not subject to premarket approval,” said Susan Laine from the FDA Office of Media Affairs. “510(k) (premarket notification) to FDA is required at least 90 days before marketing, unless the device is exempt from 510(k) requirements.”
PMA is the most stringent type of device-marketing application required by the FDA. Unlike premarket notification, PMA is based on a determination by the FDA that the PMA contains sufficient valid scientific evidence to provide reasonable assurance that the device is safe and effective for its intended use or uses.
Which regulatory requirement is needed depends on the class of medical device. For example, most Class I devices are exempt from 510(k) premarket notification; most Class II devices require the 510(k); and most Class III devices require PMA.
The FDA cannot provide statistics on the number of companies that fail or give up during the approval process. Laine noted that the clearance and approval process is interactive and not always linear, with some premarket submissions needing additional information from manufacturers before a review can go forward.
“Also, companies may make business-related decisions independent of FDA that result in applications being withdrawn or not completed,” she said. “So, it is difficult to quantify how many premarket applications are started but not completed, and why that happens. However, when a company submits an application that is lacking necessary information, they receive written feedback from the FDA outlining the deficiencies in their application so they know what is expected to make their submission complete.”
One effective way that the FDA provides manufacturers with some predictability is in outlining its thinking about an issue or process and providing a clear understanding of expectations in a certain area. The FDA has issued a number of guidance documents in the areas of benefit-risk determinations, presubmissions interaction and standards acceptance.
The FDA’s best advice to companies is to come to them early when they are still developing a medical device. “When companies work FDA’s expectations into the medical device development process, they are less likely to experience major surprises during the review process,” Laine said.
Top links for information on medical device approval
International overview
• Worldwide health care expenditure: http://www.emergogroup.com/resources/infographics
• Medical device regulations by country: http://www.emergogroup.com/resources/worldwide-medical-device-regulations
• Videos and webinars: http://www.emergogroup.com/resources/videos
• Medical device market information: http://www.emergogroup.com/resouces/medical-device-market-information
CE marking in the UK and Europe
• Medicines and Healthcare products Regulatory Agency (MHRA): http://www.mhra.gov.uk/howweregulate/index.htm
• Association of British Healthcare Industries (ABHI): http://www.abhi.org.uk
• Conformance Ltd.: http://www.conformance.co.uk/adirectives/doku.php?id=medical
• European Commission Regulatory Framework: http://ec.europa.eu/health/medical-devices/regulatory-framework/index_en.htm
• Emergo Group white paper: http://www.emergogroup.com/resources/white-paper-europe-ce-marking
FDA approval in the US
• Overview of device regulation: http://www.fda.gov/medicaldevices/deviceregulationandguidance/overview/default.htm
• Product Classification Database: http://www.fda.gov/medicaldevices/deviceregulation andguidance/overview/classifyyourdevice/default.htm
• Presubmissions assistance for a premarket application: http://www.fda.gov/medicalde
vices/deviceregulationandguidance/guidancedocuments/ucm310375.htm
• US FDA regulatory process for medical devices (7-minute video): http://www.emergogroup.com/resources/videos-us-fda-regulatory-process
Steps to approval
The medical device regulations process varies by market and could occupy an entire book; however, all markets follow the same basic steps:
• Determine if your product qualifies as a “medical device.”
• Determine its classification according to national laws.
• Determine the path to regulatory approval.
• Conduct clinical trials (mostly high-risk devices only).
• Prepare a technical file or dossier with detailed technical information about the device.
• Appoint a local regulatory representative if you have no office in the market you plan to enter. Most countries have this requirement.
• Comply with local quality management system requirements. Applies in the US, EU and many markets.
• Submit your technical file and wait for approval from the authorities.
Step-by-step details of the process for each market can be found at http://www.emergogroup.com/resources/regulatory-process-charts.
Top 25 Countries
% of world health expenditures
1. United States: 40.1%
2. Japan: 8%
3. Germany: 5.9%
4. France: 4.7%
5. China: 4.6%
6. United Kingdom: 3.4%
7. Italy: 3%
8. Brazil: 3%
9. Canada: 2.8%
10. Spain: 2.1%
11. Australia: 1.6%
12. Netherlands: 1.4%
13. Russia: 1.2%
14. South Korea: 1.1%
15. Mexico: 1.1%
16. India: 1%
17. Switzerland: 0.9%
18. Belgium: 0.8%
19. Turkey: 0.8%
20. Sweden: 0.7%
21. Austria: 0.6%
22. Norway: 0.6%
23. Denmark: 0.6%
24. Poland: 0.5%
25. South Africa: 0.5%
The 25 countries listed above account for more than 90% of global health expenditures.