Alec De Grand and Samuel Bonfig, Olympus
Optical microscopy has advanced greatly over the past decade. Here’s how to select the right instrument for any particular application.
Selecting the right optical microscopy system relies on a variety of factors: whether the instrument will be used by one researcher or a core facility, how much flexibility is needed for future changes, expected budget, ease of training, footprint and location in the lab, ability to interface with existing equipment, and much more.
Imaging depth is one of the most important selection considerations. All imaging techniques seek to maximize signal and decrease noise, but some biological processes occur at the surface, where cell membranes interact with their environments, while others happen deep beneath the surface. Specimen depth is one of the most complex and important aspects of evaluating instrument options, and experiments at every depth have their own challenges. The deeper the specimen of interest is, the greater the challenges. Light scatter, absorption, background signal, difficulty in collecting enough photons at the detector, and refractive index differences present a variety of hurdles; from the shallowest to deepest depths, every type of imaging can be optimized with a system designed to meet its special challenges.
TIRF: 0-200 µm beneath specimen surface
For imaging just below the surface of a specimen, many scientists choose total internal reflection fluorescence (TIRF) imaging (Figure 1). A TIRF system typically includes a research-grade fluorescence microscope, together with a specialized optics and laser. TIRF is an optical phenomenon used to observe events occurring at or just beneath boundaries, such as where the cell membrane touches the coverslip. When light strikes the interface between materials with different refractive indices, the light entering at an angle greater than the critical angle undergoes total reflection. Beyond the angle of total reflection, the electromagnetic field of the incoming/reflected light extends into the Z axis, but the strength of this evanescent wave decreases exponentially as it extends through the second medium.
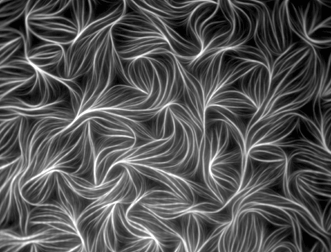
Figure 1. Fluorescently labeled actin from chicken muscle. Image captured with TIRF microscopy by Dennis Breitsprecher of Hannover Medical School, Institute of Biophysical Chemistry, in Hannover, Germany. The image received an honorable mention in the 2010 Olympus BioScapes Digital Imaging Competition.
TIRF microscopy is a powerful way of elucidating processes occurring at the cell surface, including adhesion, hormone binding, neurotransmitter secretion and membrane dynamics. The shallow penetration depth of the evanescent wave allows only those fluorophores near the surface of a specimen to become excited, creating an extremely thin optical section. Fluorescence outside the evanescent field is minimal, providing high-contrast images with good signal-to-noise ratios at the right depth. TIRF requires high-numerical-aperture (NA) objectives; however, even with the highest-NA objectives, such as a 1.7-NA 100X, TIRF imaging is limited to about 200 µm in the Z axis.
Wide-field fluorescence: 0-700 µm beneath specimen surface
Researchers seeking to view nuclei, stem cells, certain brain slices, neuron signaling, DNA and chromosomes often use wide-field epifluorescence microscopy. In fluorescence imaging, the specimen is illuminated by a specific wavelength of light that is absorbed by fluorophores attached to it. The fluorophores trigger the emission of longer wavelengths of light, which are then amplified and detected. Because both the reflected excitatory light and emitted fluorescence are collected along the same optical path, epifluorescence produces a high signal-to-noise ratio.
For the researcher interested in imaging specimens at mid-depth, wide-field fluorescence microscopy can offer beautiful images and excellent data at a relatively low price point (Figure 2). Because individual fluorophores (fluorescent probes) cause light to be emitted in an extraordinary variety of colors, users often have substantial control over the imaging process. Some systems are sensitive enough to enable detection of specific fluorophores at low concentrations in complex mixtures over time with single-molecule resolution. It is relatively simple to create multicolor images by combining scans created with different fluorophores.
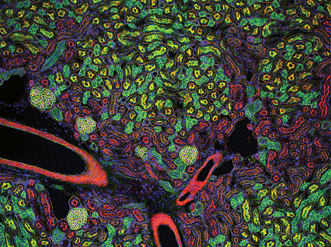
Figure 2. Mouse kidney cryostat section. Nuclei (blue), glomeruli (yellow-green), convoluted tubules (green) and proximal tubules (red) are visible. Gilles Grondin, University of Sherbrooke in Quebec, Canada. The 100× fluorescence image received an honorable mention in the 2006 Olympus BioScapes Digital Imaging Competition.
However, it sometimes can be challenging to generate enough fluorescence emission. Biological specimens typically emit fluorescence signals three to six orders of magnitude less than the illumination signal, making it challenging to detect molecules of interest, particularly at greater depths. Additionally, repeated exposure of the specimen to light leads to photobleaching, which reduces fluorescence emission and degrades image contrast very quickly. Finally, the inability of wide-field epifluorescence to reduce background noise outside the focal plane makes it generally unsuitable below 700 µm deep.
Confocal imaging: 0-1000 µm beneath specimen surface
When live tissue samples are particularly thick and have multiple planes of interest, they can sometimes scatter so much light that very little specimen-emitted fluorescence reaches the detector. For every specimen, there is a depth at which scattering, photobleaching and noise occur at such high levels that traditional fluorescence techniques are no longer effective.
Confocal microscopes are used for three-dimensional fluorescence imaging within living specimens because they remove out-of-focus light from outside the focal plane and permit thin optical sectioning (Figure 3). As the powerhouses of many life science research laboratories, confocal systems allow scientists to study life processes in real time as they occur. Three-dimensional views are created by
building stacks of individual sections from each optical slice collected at sequential Z axis locations.
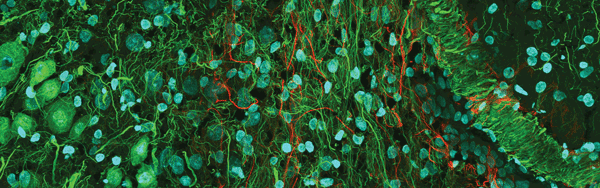
Figure 3. Confocal mouse brain, 1 × 3 portion of a 3 × 3 stitched image. The sample was labeled for DAPI (blue), actin (green) and neurofilament (red). Image courtesy of Olympus America Inc.
But confocal systems have limitations. Because the excitation light generates fluorescence throughout the entire depth of the sample, confocal microscopes can damage or bleach the entire volume of the specimen. Also, the deeper the region of interest is, the harder it is to collect the emitted signal. Light must travel from the sample, through the microscope’s optical system and pass through a small confocal aperture – typically a slit or pinhole – to the detector. Any deflection or scattering of the emitted fluorescence in the sample results in its rejection by the confocal aperture, meaning that photons traveling from deep within the specimen experience high levels of scattering and rejection.
Multiphoton microscopy: 0-2000 µm below specimen surface
Multiphoton microscope systems allow imaging at much deeper levels than their confocal cousins, typically up to about 2000 µm below the surface. They rely on much longer wavelengths of light to excite fluorescence specimens and scatter less illumination as they scan through tissue. They also are less harmful to living cells.
In multiphoton imaging, two or more long-wavelength, lower-energy excitation photons are absorbed simultaneously by a single fluorophore. When this happens, the two photons combine their energies to excite the fluorophore and stimulate fluorescence as if they were a single high-energy (low-wavelength) excitation photon. In contrast to confocal microscopy, where fluorescence is generated throughout the entire specimen volume, multiphoton fluorescence excitation occurs only at the plane of focus. As a result, there is less phototoxicity and photobleaching, and tissue can be imaged for much longer periods of time.
Multiphoton microscopy also allows the investigation of ultrathick brain slices, eye tissue, developing embryos and other living tissue that is so thick or dense that it cannot be penetrated with conventional imaging techniques (Figure 4). Multiphoton methodologies are especially useful for neuroscientists, cell biologists and other researchers who wish to study dynamic processes over time in living cells and tissues while retaining viable specimens for as long as possible – over hours, days or even weeks. The rapid pulsing of lasers allows fast functions like Ca+ signaling along neurons to be imaged in real time as parts of the brain are stimulated.
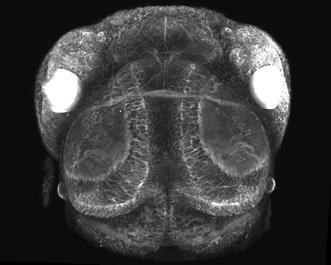
Figure 4. Multiphoton image of a living zebra fish: CFP and YFP expression during embryonic eye devolvement (25×, 1.05-NA objective). Image courtesy of Philip Williams and Rachel Wong, University of Washington.
Multiphoton with clearing: 0-8000 µm beneath the specimen surface
Until recently, the idea of imaging beyond a millimeter into fixed tissue was unthinkable, but recent developments in clearing techniques have allowed researchers to image up to 8000 nm (8 mm) deep using fixed specimens. New systems use multiphoton microscopes, ultralong-working-distance optics and proprietary clearing reagents to render specimens transparent to facilitate the study of deep, contiguous tissue within the brain and other dense organs.
These clearing methodologies, such as Scale and Clarity, have several key advantages. First, they allow visualization of connections even through the dense, light-scattering tissue of the brain. In addition, they vastly reduce the need to slice the specimen, which damages the tissue at the slice and makes it difficult to determine the thread of individual neural structures amid the tangled web of brain connections.
In August 2011, Dr. Atsushi Miyawaki and his colleagues at the Riken Brain Science Institute in Japan published data1 indicating that a clearing agent they created, together with the specially developed 25X multiphoton Olympus Scaleview objective, allowed viewing several times deeper into contiguous tissue than had ever before been possible – up to 8 mm deep (Figure 5). Correction collars allowed for optimization of lens focus within even the deepest tissue.
Figure 5. With image-clearing techniques such as Scaleview, researchers can image up to 8 mm deep into dense brain tissue without slicing. Raw image data courtesy of Hiroshi Harna, Hiroshi Kurokawa and Atsushi Miyawaki of the Riken Brain Science Institute’s Laboratory for Cell Function Dynamics in Japan.
Clarity,2 which comes out of Dr. Karl Deisseroth’s laboratory at Stanford University, is another prominent clearing technique. It involves creating a stable tissue-hydrogel hybrid out of thick biological tissue without changing the tissue’s original structure. Clarity has the potential to help researchers extract important neurobiological information from deep, intact tissue.
Both of these deep-imaging techniques have enormous potential to change the face of neuroscience. However, at this time, they may only be used with fixed tissue.
Superresolution: enhancing resolution at any depth
“Superresolution” describes a variety of imaging techniques that can improve the lateral (X,Y) resolution of a microscope beyond the theoretical diffraction limit of the optical system. Many superresolution techniques employ sequential activation and time-resolved localization of photoswitchable fluorophores to create images. Superresolution systems have benefits for axial (Z axis, or depth) resolution as well. Structured illumination microscopy, for instance, can provide for a Z resolution as great as 300 nm.3
Although superresolution methods have enormous potential for helping researchers image phenomena they could never see before, they still have limitations. Most require expensive hardware such as special probes for photoswitching and additional laser systems for localization. They also involve very particular sample preparations, making the methodologies difficult to implement in some settings. In addition, superresolution is highly dependent on mathematical models, making the computational, storage and upkeep requirements very high. Several companies now offer affordable software-based options that can provide many of the benefits of traditional superresolution.
Advice to users
Imaging can be a challenge at any depth within a specimen. But optical microscopy has made great strides in imaging over the past decade, from capturing phenomena at the surface down to ever-increasing depths. The key to selecting the right instrument for any particular application is to keep the following factors in mind: who will use it, current experimental parameters and laboratory environment, future needs and overall resources available. A trusted colleague or applications representative may provide useful information on applicability, footprint, cost of ownership over time, flexibility for upgrading, ease of training and more. Any experiment can be enhanced by making the right choice regarding imaging methodology and equipment.
Meet the authors
Alec De Grand and Samuel Bonfig are both employees of Olympus Scientific Solutions Americas in Waltham, Mass.; email: [email protected] and [email protected].
References
1. H. Hama et al (2011). SCALE: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat Neurosci, Vol. 14, pp. 1481-1488.
2. K. Chung and K. Deisseroth (2013). CLARITY for mapping the nervous system. Nat Methods, Vol. 10, pp. 508-513.
3. L. Shao et al (2012). Interferometer-based structured-illumination microscopy utilizing complementary phase relationship through constructive and destructive image detection by two cameras. J Microsc, Vol. 246, pp. 229-236.
4. E. Betzig et al (2006). Imaging intracellular fluorescent proteins at nanometer resolution. Science, Vol. 313, p. 1642.
5. J.A. Conchello and J.W. Lichtman (2005). Optical sectioning microscopy. Nat Methods, Vol. 2, pp. 920-931.
6. W. Denk et al (1990). Two-photon laser scanning fluorescence microscopy. Science, Vol. 248, pp. 73-76.
7. M.G.L. Gustafsson (2005). Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. P Natl Acad Sci USA, Vol. 102, pp. 13081-13086.
8. S.W. Hell and J. Wichmann (1994). Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt Lett, Vol. 19, pp. 780-782.
9. D.B. Murphy and M.W. Davidson (2013). Fundamentals of Light Microscopy and Electronic Imaging. Second Edition. Wiley-Blackwell.
10. J. Pawley, editor (2006). Handbook of Biological Confocal Microscopy. Third Edition. Springer.
11. M.J. Rust et al (2006). Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods, Vol. 3, pp. 793-796.
12. L. Shao et al (2011). Super-resolution 3D microscopy of live whole cells using structured illumination. Nat Methods, Vol. 8, pp. 1044-1046.
13. N.L. Thompson (1981). Measuring surface dynamics of biomolecules by total internal reflection fluorescence with photobleaching recovery or correlation spectroscopy. Biophys J, Vol. 33, pp. 435-454.
14. R. Tomer (2014). Advanced CLARITY for rapid and high-resolution imaging of intact tissues. Nat Protoc, Vol. 9, pp. 1682-1697.